Вам также может понравиться
- Genetica BacterianaДокумент22 страницыGenetica Bacterianaaxel_gutierrez_11Оценок пока нет
- Recombinacion Genetica en CepasДокумент5 страницRecombinacion Genetica en CepasFreddy Diaz GarciaОценок пока нет
- Práctica Molecular PlásmidosДокумент7 страницPráctica Molecular PlásmidosAngelica HgОценок пока нет
- Trabajo Biologia Celular Expresión Recombinante en E. ColiДокумент6 страницTrabajo Biologia Celular Expresión Recombinante en E. Coliluisa Maria RendonОценок пока нет
- Seminario 3Документ11 страницSeminario 3Mariana AgustinaОценок пока нет
- Genética Bacteriana PDFДокумент40 страницGenética Bacteriana PDFLuz Mary Montes RamírezОценок пока нет
- Práctica 2. TransformaciónДокумент1 страницаPráctica 2. TransformaciónYumari Martinez GalvanОценок пока нет
- Genética Bacteriana..Документ8 страницGenética Bacteriana..Vicente Huanca CanaviriОценок пока нет
- Genética BacterianaДокумент2 страницыGenética BacterianaFernanda SotoОценок пока нет
- Expresion Genetica en BacteriasДокумент17 страницExpresion Genetica en BacteriasFer RodriguezОценок пока нет
- Genetic A Bacterian AДокумент22 страницыGenetic A Bacterian ADieiKonCaiCedОценок пока нет
- Manipulación de La Expresión Génica en BacteriasДокумент16 страницManipulación de La Expresión Génica en BacteriasAna Julia TicchiОценок пока нет
- ClonaciónДокумент9 страницClonaciónMelissa GalanОценок пока нет
- Informe de Genética BacterianaДокумент8 страницInforme de Genética BacterianaOwen Harvey Quispe CasillaОценок пока нет
- Trabajo de GeneticaДокумент5 страницTrabajo de GeneticaYucelis Paola Herazo SalgadoОценок пока нет
- Transformación GeneticaДокумент4 страницыTransformación GeneticaEduardo Cruz FОценок пока нет
- Vectores de ClonaciónДокумент3 страницыVectores de Clonaciónselenelemus259Оценок пока нет
- Conjugación ProcariotaДокумент4 страницыConjugación ProcariotaAntonio TreminiОценок пока нет
- Practica 7. Clonacion.Документ5 страницPractica 7. Clonacion.ARLEN SCOLY MAMANI ONSIHUAYОценок пока нет
- Clonaje Del ADNДокумент4 страницыClonaje Del ADNSeries HugoОценок пока нет
- Cuestionario TransformaciónДокумент2 страницыCuestionario TransformaciónMarianaMtz1995Оценок пока нет
- PlasmidosДокумент5 страницPlasmidosKarhito BMОценок пока нет
- Anticuerpos MonoclonalesДокумент27 страницAnticuerpos MonoclonalesSendar Nery100% (1)
- Resumen 5 MicroДокумент11 страницResumen 5 Microhv66580Оценок пока нет
- Plasmidos y TransposonesДокумент22 страницыPlasmidos y TransposonesIndira BedriñanaОценок пока нет
- Transformación E. Coli Con GFPДокумент11 страницTransformación E. Coli Con GFPViktoryia RybakovaОценок пока нет
- Genetica BacterianaДокумент19 страницGenetica BacterianaJoel Ramirez PadillaОценок пока нет
- Control Genético en ProcariotasДокумент9 страницControl Genético en ProcariotasleomelthormeОценок пока нет
- Genetica BacterianaДокумент32 страницыGenetica BacterianaLeonely Delgado C.Оценок пока нет
- CUESTIONARIO PRACTICA 3 LMFFL - Erick Humberto Salgado Bahena PDFДокумент9 страницCUESTIONARIO PRACTICA 3 LMFFL - Erick Humberto Salgado Bahena PDFErick SalgadoОценок пока нет
- Genética BacterianaДокумент29 страницGenética BacterianaLuisОценок пока нет
- Recombinación BacterianaДокумент23 страницыRecombinación BacterianaStefany Beni Ruiz50% (2)
- Transformación BacterianaДокумент8 страницTransformación BacterianaFrancisco Santander RomeroОценок пока нет
- Ingenieria GeneticaДокумент23 страницыIngenieria GeneticaAraceli CóceresОценок пока нет
- Recombinacion GeneticaДокумент3 страницыRecombinacion GeneticaAntony SIESQUEN DIAZОценок пока нет
- ADN Recombinante II-1Документ3 страницыADN Recombinante II-1MARIO CRISPIN HERNANDEZ ALVAREZОценок пока нет
- Genetica y Replicacion ViralДокумент13 страницGenetica y Replicacion ViralMaritza Velasquez TОценок пока нет
- Plasmidos de MolecularДокумент7 страницPlasmidos de Molecularandres zuluagaОценок пока нет
- Genoma ProcariotaДокумент14 страницGenoma ProcariotaAbel MooreОценок пока нет
- Proteínas de Interés FarmaceuticoДокумент17 страницProteínas de Interés FarmaceuticoPaulii H ArmenttaОценок пока нет
- Ingeniería Genética en Organismos EucarióticosДокумент32 страницыIngeniería Genética en Organismos EucarióticosRaquel Pérez FernándezОценок пока нет
- PIOLINДокумент21 страницаPIOLINerika ccala choqueОценок пока нет
- Microbiología - Taller PDFДокумент6 страницMicrobiología - Taller PDFvaluОценок пока нет
- Taller Ingeniería GenéticaДокумент4 страницыTaller Ingeniería Genéticamarcela velascoОценок пока нет
- Resumen InmunoДокумент13 страницResumen InmunoLaura Rosa Guerra SalasОценок пока нет
- Clonacion y PCRДокумент2 страницыClonacion y PCRCamila ÁvilaОценок пока нет
- Clonación en Hongos. FaltaДокумент47 страницClonación en Hongos. FaltaMau Kriss FiallosОценок пока нет
- Genetica BacterianaДокумент24 страницыGenetica BacterianaYanny GarciaaОценок пока нет
- Ingeniería Genética en Organismos EucarióticosДокумент33 страницыIngeniería Genética en Organismos EucarióticosRaquel Pérez FernándezОценок пока нет
- PlasmidosДокумент10 страницPlasmidosAnggeLОценок пока нет
- 2015lmffl Pra4 Eq1 6b RevlutДокумент23 страницы2015lmffl Pra4 Eq1 6b RevlutMayren Román100% (1)
- Clonacion de BacteriasДокумент5 страницClonacion de BacteriasEdwin Amador LinoОценок пока нет
- E - Coli Silvestre y MejoradaДокумент4 страницыE - Coli Silvestre y MejoradaSantiago ValenciaОценок пока нет
- TransfecciónДокумент5 страницTransfecciónFernando Crespo OrellanaОценок пока нет
- 5.-Genetica Microbiana 2021Документ51 страница5.-Genetica Microbiana 2021Esmeralda HernandézОценок пока нет
- Proteinas Recombinantes PDFДокумент3 страницыProteinas Recombinantes PDFIndira Romero HerediaОценок пока нет
- Rejuvenecer Con El Plasma Sanguíneo De Los JóvenesОт EverandRejuvenecer Con El Plasma Sanguíneo De Los JóvenesРейтинг: 5 из 5 звезд5/5 (1)
- Acceso a Universidad para Mayores de 25 años. Biología 2013-2017.: Solucionario Pruebas 2013-2017От EverandAcceso a Universidad para Mayores de 25 años. Biología 2013-2017.: Solucionario Pruebas 2013-2017Оценок пока нет
- Introducción a la Biología: RESÚMENES UNIVERSITARIOSОт EverandIntroducción a la Biología: RESÚMENES UNIVERSITARIOSРейтинг: 5 из 5 звезд5/5 (1)
- Extraccion ARNДокумент35 страницExtraccion ARNGabriela BuendiaОценок пока нет
- PrecipitacionДокумент4 страницыPrecipitacionGabriela Buendia100% (1)
- Cinetica y Crecimiento Bacteriano PDFДокумент2 страницыCinetica y Crecimiento Bacteriano PDFGabriela BuendiaОценок пока нет
- Longitudes EquivalentesДокумент1 страницаLongitudes EquivalentesGabriela BuendiaОценок пока нет
- Ruptura CelularДокумент31 страницаRuptura CelularGabriela BuendiaОценок пока нет
- Solucion Problema 4 IntercambiadoresДокумент4 страницыSolucion Problema 4 IntercambiadoresGabriela BuendiaОценок пока нет
- Cálculo de MuestraДокумент9 страницCálculo de MuestraluisccorralОценок пока нет
- Bioquimica Informe Sem 1Документ18 страницBioquimica Informe Sem 1Vivian Sotomayor FloresОценок пока нет
- Fase 3. Tejido Adiposo e InflamaciónДокумент1 страницаFase 3. Tejido Adiposo e InflamaciónAylin Almeyda GonzalezОценок пока нет
- 95-Texto Del Artículo-147-1-10-20200131Документ6 страниц95-Texto Del Artículo-147-1-10-20200131Laura HerreraОценок пока нет
- PRACTICA N 5 BCI 2020 Aminoácidos y ProteínasДокумент13 страницPRACTICA N 5 BCI 2020 Aminoácidos y ProteínasDavid Carlos Bertolotto Huamaní100% (1)
- Trastornos Genéticos Por Trastornos en ReceptoresДокумент31 страницаTrastornos Genéticos Por Trastornos en ReceptoresBrayan EstradaОценок пока нет
- FARMACOLOGÍASEM4Документ7 страницFARMACOLOGÍASEM4Carlos GuzmánОценок пока нет
- Cuadro SinopticoДокумент1 страницаCuadro SinopticoErick polancoОценок пока нет
- Tabla Funciones OrgánulosДокумент4 страницыTabla Funciones OrgánulosMiriam FournonОценок пока нет
- Niveles de Organización de La VidaДокумент3 страницыNiveles de Organización de La VidaJosue PortilloОценок пока нет
- PremedicinaДокумент6 страницPremedicinaLaura ZetinaОценок пока нет
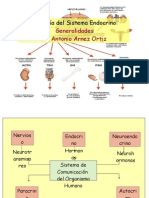
- Sistema Endocrino - GeneralidadesДокумент26 страницSistema Endocrino - GeneralidadesJosé Eduardo Pinheiro MatosОценок пока нет
- SX de Mala Absorción IntestinalДокумент24 страницыSX de Mala Absorción Intestinaldany2515 QuinteroОценок пока нет
- Examen NДокумент3 страницыExamen Nmaxwim1984Оценок пока нет
- CAPÍTULO 59 - Anatomía y Fisiología de PáncreasДокумент12 страницCAPÍTULO 59 - Anatomía y Fisiología de PáncreasBelen BonillaОценок пока нет
- Parásito-Foro 10 - Identificación de TremátodosДокумент7 страницParásito-Foro 10 - Identificación de TremátodosPierina SivinchaОценок пока нет
- BIOQUIMICA EVALUACION FinalДокумент14 страницBIOQUIMICA EVALUACION FinalLuis Eduardo GomezОценок пока нет
- Sesión 09 Fotosintesis y FasesДокумент13 страницSesión 09 Fotosintesis y Fasesazulmarino78Оценок пока нет
- Práctica 4 Integridad ADN Mediante ElectroforesisДокумент3 страницыPráctica 4 Integridad ADN Mediante ElectroforesisSuelen NunesОценок пока нет
- Informe ProteinasДокумент8 страницInforme ProteinasMacky MacОценок пока нет
- "Demostración de La Fermentación LácticaДокумент11 страниц"Demostración de La Fermentación LácticaCINTHIA DEL PILAR TASILLA TANTAОценок пока нет
- Proteínas - MonografiasДокумент4 страницыProteínas - Monografiasana cruzОценок пока нет
- Autoevaluación Módulo 1Документ4 страницыAutoevaluación Módulo 1Carol Mora PlaОценок пока нет
- Amilasa SalivalДокумент4 страницыAmilasa SalivalRenato Omar Bustamante SilvaОценок пока нет
- Proteínas UNAHДокумент7 страницProteínas UNAHAlejandra FunezОценок пока нет
- Toxicos Naturales - AntinutrientesДокумент34 страницыToxicos Naturales - AntinutrientesJomayra Requena100% (2)
- Resumen Informe #11Документ4 страницыResumen Informe #11Dario Quimis SantanaОценок пока нет
- Wuolah Free TEMA 6 Variacion Del GenomaДокумент7 страницWuolah Free TEMA 6 Variacion Del GenomapaulaОценок пока нет
- CelulaДокумент120 страницCelulacarlos100% (2)
- Ensayo de Rosalind FranklinДокумент2 страницыEnsayo de Rosalind FranklinVini GallegosОценок пока нет
- Semana 5 - BiologíaДокумент3 страницыSemana 5 - BiologíaCÍRCULO DE ESTUDIO MATEMÁTICO GAUSSОценок пока нет