Вам также может понравиться
- Enfermedades sistémicas en el consultorio odontológico: Conocimientos básicos odontológicos, #3От EverandEnfermedades sistémicas en el consultorio odontológico: Conocimientos básicos odontológicos, #3Рейтинг: 5 из 5 звезд5/5 (1)
- 2 Introduccion Al Estudio Del Paciente HematologicoДокумент9 страниц2 Introduccion Al Estudio Del Paciente HematologicoAldo100% (1)
- Aplasia MedularДокумент6 страницAplasia MedularDavid Ross50% (2)
- Aplasia Medular Medicina IIIДокумент28 страницAplasia Medular Medicina IIIIvania Claudeth Salinas FernandezОценок пока нет
- Síndromes de Fracaso Medular: Anemia Aplásica, Eritroblastopenias Selectivas y Anemias DiseritropoyéticasДокумент7 страницSíndromes de Fracaso Medular: Anemia Aplásica, Eritroblastopenias Selectivas y Anemias DiseritropoyéticasKurosaki IchigoОценок пока нет
- Anemia HipoplasicaДокумент19 страницAnemia HipoplasicaMelisa FlorianОценок пока нет
- Anemia Aplasica TrabajoДокумент12 страницAnemia Aplasica TrabajoIngrid Briggitte Pacherres OlayaОценок пока нет
- Anemia HipoplasicaДокумент4 страницыAnemia HipoplasicaIris Miranda100% (1)
- Aplasia MedularДокумент43 страницыAplasia MedularGenesis Nicole Zapata NavarreteОценок пока нет
- Fallo MedularДокумент28 страницFallo MedularToto Alejandro100% (1)
- Medicine - Sindromes de Fracaso MedularДокумент7 страницMedicine - Sindromes de Fracaso MedularJorge Ricardo Reyes RoelОценок пока нет
- Insuficiencia MedularДокумент33 страницыInsuficiencia MedularFranco SáezОценок пока нет
- C01 NeutropeniaДокумент8 страницC01 Neutropeniajuan ugazОценок пока нет
- PancitopeniaДокумент17 страницPancitopeniaBenjamin Torres Octavo100% (1)
- HC y EF en El Paciente HematologicoДокумент12 страницHC y EF en El Paciente HematologicoSuleivi LoraОценок пока нет
- Amiloidosis RenalДокумент8 страницAmiloidosis RenalRodrigoAprendeBienОценок пока нет
- 08 Enf AutoinmunesДокумент33 страницы08 Enf AutoinmunesFrancis Yaringaño TorresОценок пока нет
- Falla MedularДокумент5 страницFalla MedularMARTHA ISABEL CHUNG REYESОценок пока нет
- 03.018 Protocolo Diagnóstico de La NeutropeniaДокумент4 страницы03.018 Protocolo Diagnóstico de La NeutropeniaGustavo Araujo100% (1)
- Anemia AplasicaДокумент26 страницAnemia Aplasicachizko14Оценок пока нет
- Aplasia MedularДокумент4 страницыAplasia MedularClemencia Alvarez VillaОценок пока нет
- Nefropatia LUpica.Документ16 страницNefropatia LUpica.Jacqueline MossoОценок пока нет
- PancitopeniaДокумент14 страницPancitopeniaroge-leon50% (2)
- Alteraciones de Las PlaquetasДокумент10 страницAlteraciones de Las PlaquetasPriscila Ribotti SheenОценок пока нет
- 18 Sindrome Nefrotico PDFДокумент19 страниц18 Sindrome Nefrotico PDFjuliogasteloОценок пока нет
- Clase de Lupus Eritematosos Sistemico 2007Документ52 страницыClase de Lupus Eritematosos Sistemico 2007Alexiran SaavedraОценок пока нет
- Anemia AplasicaДокумент2 страницыAnemia AplasicaCECILIA MARCELA CASTRO SARABIAОценок пока нет
- Linfoma No HodgkinДокумент68 страницLinfoma No HodgkinEdithRuelasRodriguezОценок пока нет
- Leucemia en PediatríaДокумент43 страницыLeucemia en PediatríaJuan Pablo Henríquez EscuderoОценок пока нет
- 4 - Lupus Eritematoso SistémicoДокумент8 страниц4 - Lupus Eritematoso SistémicoFVPОценок пока нет
- Anemia AplásicaДокумент8 страницAnemia AplásicaAurora GonzalezОценок пока нет
- Aplacia MedularДокумент4 страницыAplacia Medularwandher monteroОценок пока нет
- Lupus Eritematoso SistémicoДокумент6 страницLupus Eritematoso SistémicoGonzalo VeizagaОценок пока нет
- Lupus Eritematoso SistémicoДокумент2 страницыLupus Eritematoso SistémicotrofincrisОценок пока нет
- Síndromes MielodisplásicosДокумент11 страницSíndromes MielodisplásicosGeraldineAnabellaОценок пока нет
- Resumen de Catedra de HEMATO 2Документ26 страницResumen de Catedra de HEMATO 2Catalina MoyaОценок пока нет
- Marco TeoricoДокумент11 страницMarco TeoricomonicammarmolejoОценок пока нет
- Lupus Eritematoso Sistemico PediatricoДокумент6 страницLupus Eritematoso Sistemico PediatricoD Ivette Santiago FloresОценок пока нет
- Síndrome Antifosfolípido: ResumenДокумент14 страницSíndrome Antifosfolípido: ResumenJesus Del RioОценок пока нет
- Anemias Por Defecto de ProliferaciónДокумент9 страницAnemias Por Defecto de ProliferaciónpatriciaОценок пока нет
- Sindrome Antifosfolipido 2017Документ44 страницыSindrome Antifosfolipido 2017Paz Vida100% (1)
- Clase 5. Anemia AplasicaДокумент12 страницClase 5. Anemia AplasicaYesse CruzОценок пока нет
- Mielofibrosis PrimariaДокумент3 страницыMielofibrosis PrimariaXime G. AОценок пока нет
- Leucemias AgudasssДокумент5 страницLeucemias AgudasssAlessandra PisanoОценок пока нет
- PC Atresia Esofágica 2 New 2Документ42 страницыPC Atresia Esofágica 2 New 2Ana LauОценок пока нет
- Sindrome Urémico HemolíticoДокумент9 страницSindrome Urémico HemolíticoluisinothОценок пока нет
- Tema 5. Anemia AplasicaДокумент4 страницыTema 5. Anemia Aplasicajuliodelgadosec03Оценок пока нет
- Sindromes de Falla Medular - PPTX 1Документ58 страницSindromes de Falla Medular - PPTX 1Jeanpierre VillarrubiaОценок пока нет
- HEMATOLOGIAДокумент205 страницHEMATOLOGIAOmar Morales100% (2)
- Absorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleОт EverandAbsorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleОценок пока нет
- Comprendiendo El Alzhéimer: 7 mitos y verdades que debes conocerОт EverandComprendiendo El Alzhéimer: 7 mitos y verdades que debes conocerРейтинг: 5 из 5 звезд5/5 (1)
- Fundamentos del diagnóstico y tratamiento del cáncer en adultos: Una aproximación inicial para el médico no especialista en cáncerОт EverandFundamentos del diagnóstico y tratamiento del cáncer en adultos: Una aproximación inicial para el médico no especialista en cáncerОценок пока нет
- Urgencias médicas en el consultorio odontológico: Conocimientos básicos odontológicos, #2От EverandUrgencias médicas en el consultorio odontológico: Conocimientos básicos odontológicos, #2Рейтинг: 5 из 5 звезд5/5 (1)
- Errores innatos en el metabolismo: Un abordaje integral del diagnóstico al tratamientoОт EverandErrores innatos en el metabolismo: Un abordaje integral del diagnóstico al tratamientoОценок пока нет
- Bases Anatomopatológicas De La Enfermedad Quirúrgica: Tomo IiОт EverandBases Anatomopatológicas De La Enfermedad Quirúrgica: Tomo IiОценок пока нет
- Profilaxis Encefalopatia HepaticaДокумент3 страницыProfilaxis Encefalopatia HepaticaManuel MoralesОценок пока нет
- CrossДокумент2 страницыCrossManuel MoralesОценок пока нет
- Consenso POSTRCPДокумент20 страницConsenso POSTRCPManuel MoralesОценок пока нет
- Biblia de RadiologiaДокумент1 430 страницBiblia de RadiologiaAlex Daza100% (3)
- Apn Reenfocada 1era. Atencion IntegralДокумент27 страницApn Reenfocada 1era. Atencion IntegralManuel MoralesОценок пока нет
- Brucelosis NeonatalДокумент3 страницыBrucelosis NeonatalManuel MoralesОценок пока нет
- Bursa Sub AcromialДокумент3 страницыBursa Sub AcromialManuel MoralesОценок пока нет
- Manual NutriciónДокумент50 страницManual Nutriciónaliciacar100% (1)
- Examen MIR 23-01-2010 by Villa MedicДокумент38 страницExamen MIR 23-01-2010 by Villa MedicManuel Morales100% (1)
- AyurvedaДокумент43 страницыAyurvedaAna VillalbaОценок пока нет
- RedacciónДокумент22 страницыRedacciónDiego VegaОценок пока нет
- Caso KodakДокумент3 страницыCaso KodakValeria FajansОценок пока нет
- Nomenclatura de Compuestos InorgánicosДокумент8 страницNomenclatura de Compuestos InorgánicosDaniela Cardozo100% (1)
- Ciu 14Документ4 страницыCiu 14jesus_bermudez_romeroОценок пока нет
- 8.ejercicios EOQДокумент6 страниц8.ejercicios EOQBRIGITH VANESSA ORTIZ PAEZОценок пока нет
- La Efectividad de La Arteterapia para Reducir El Estrés Académico Entre Los Estudiantes Durante El Aprendizaje en Línea Es-ESДокумент10 страницLa Efectividad de La Arteterapia para Reducir El Estrés Académico Entre Los Estudiantes Durante El Aprendizaje en Línea Es-ESRobert S. Cruz NalvarteОценок пока нет
- Musumeci - ConservatoriosДокумент26 страницMusumeci - ConservatoriosLEEM FBA-UNLPОценок пока нет
- Especificaciones para Camaras Electricas de TransformacionДокумент24 страницыEspecificaciones para Camaras Electricas de TransformacionmanuelОценок пока нет
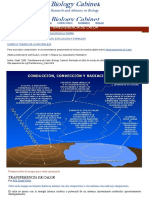
- Transferencia Del CalorДокумент8 страницTransferencia Del CalorLucas WalkerОценок пока нет
- Tarea Vi La Uapa Como Modalidad Educativa A Distancia.Документ3 страницыTarea Vi La Uapa Como Modalidad Educativa A Distancia.Orquidea FriasОценок пока нет
- Diapositivas de AlgebraДокумент34 страницыDiapositivas de AlgebraRaul B. ReyesОценок пока нет
- La Historia ClínicaДокумент15 страницLa Historia ClínicaFranco ZapataОценок пока нет
- Nemesis Serie2way Installguide EliteДокумент2 страницыNemesis Serie2way Installguide EliteJohnОценок пока нет
- Ei Cibao en SDДокумент3 страницыEi Cibao en SDVictoria HerasmeОценок пока нет
- Comput A Bili DadДокумент204 страницыComput A Bili DadRodrigo Románovich FiodorovichОценок пока нет
- Guia de Postulacion 2023Документ9 страницGuia de Postulacion 2023EDSAU CARDENAS CONISLLAОценок пока нет
- Informe N°006 Insumos para Cartel de ObraДокумент1 страницаInforme N°006 Insumos para Cartel de ObraMargory Inay Mori GarciaОценок пока нет
- Ponencia de Planificacion Curricular 2017Документ52 страницыPonencia de Planificacion Curricular 2017Kimberly HoustonОценок пока нет
- Practica 1 RefrigeracionДокумент9 страницPractica 1 RefrigeracionAlexa AltamiranoОценок пока нет
- La ManzanillaДокумент3 страницыLa ManzanillaLaura Perez LandaОценок пока нет
- Turismo GastronomicoДокумент10 страницTurismo GastronomicoJoahan Fernando Cruz HeleriaОценок пока нет
- Carlos HellerДокумент21 страницаCarlos HellerLuis Gustavo MartinezОценок пока нет
- Proyecto Final TepacheДокумент8 страницProyecto Final TepacheAidee Reyes100% (1)
- ISO TC251 WG4 MACAM May 2017 ES2Документ4 страницыISO TC251 WG4 MACAM May 2017 ES2Leonardo PinillaОценок пока нет
- Kaq 1ro Basico 20Документ4 страницыKaq 1ro Basico 20Marleny Boj100% (1)
- Tema 1-Introducción Al Estudio de La PresupuestaciónДокумент14 страницTema 1-Introducción Al Estudio de La PresupuestaciónAbdiel Tavárez100% (1)
- Obtención de Etanol Anhidro Desnaturalizado A Partir Del Ácido AcéticoДокумент28 страницObtención de Etanol Anhidro Desnaturalizado A Partir Del Ácido AcéticoCristian Condori JaraОценок пока нет
- Reporte de Practica 3.2.1.7 - 3.2.2.4Документ20 страницReporte de Practica 3.2.1.7 - 3.2.2.4Julie EsthefaniОценок пока нет