Вам также может понравиться
- ExplosivesДокумент78 страницExplosiveswaynefishing100% (10)
- Table of Contents for Explosives ManualДокумент25 страницTable of Contents for Explosives Manualjohn smith100% (1)
- Improvised Weapons & Munitions – U.S. Army Ultimate Handbook: How to Create Explosive Devices & Weapons from Available Materials: Propellants, Mines, Grenades, Mortars and Rockets, Small Arms Weapons and Ammunition, Fuses, Detonators and Delay MechanismsОт EverandImprovised Weapons & Munitions – U.S. Army Ultimate Handbook: How to Create Explosive Devices & Weapons from Available Materials: Propellants, Mines, Grenades, Mortars and Rockets, Small Arms Weapons and Ammunition, Fuses, Detonators and Delay MechanismsРейтинг: 4.5 из 5 звезд4.5/5 (5)
- A Collection of Pyrotechnic CompositionsДокумент75 страницA Collection of Pyrotechnic Compositionsapi-377076682% (11)
- Black Powder BasicsДокумент3 страницыBlack Powder Basicsmattfacory100% (2)
- Explosives Forum ArchiveДокумент1 254 страницыExplosives Forum Archivejohnny_cashedОценок пока нет
- The Preparatory Manual of ExplosivesДокумент380 страницThe Preparatory Manual of Explosivesmighele91% (11)
- Demystifying Explosives: Concepts in High Energy MaterialsОт EverandDemystifying Explosives: Concepts in High Energy MaterialsОценок пока нет
- The Complete Book of Explosive (Quer)Документ22 страницыThe Complete Book of Explosive (Quer)Bernhard Drescher86% (7)
- Steps For The Production of Potassium Chlorate For The Amateur ChemistДокумент18 страницSteps For The Production of Potassium Chlorate For The Amateur ChemistBitter_Armadillo0% (1)
- What Is A Propellant?Документ14 страницWhat Is A Propellant?Muhammad Irfan Malik50% (2)
- Nitro-Explosives: A Practical Treatise by Sanford, P. GeraldДокумент189 страницNitro-Explosives: A Practical Treatise by Sanford, P. GeraldGutenberg.org100% (3)
- How To Make SemtexДокумент14 страницHow To Make SemtexYaoxing Chia100% (2)
- How To Make DetonatorДокумент2 страницыHow To Make Detonatorr0d3n7100% (2)
- Chemical Wisdom - Horse Chestnuts and The Fermentation of Powerful PowdersДокумент67 страницChemical Wisdom - Horse Chestnuts and The Fermentation of Powerful PowdersPeter JansenОценок пока нет
- U.S. ARMY - Encyclopedia of Explosives and Related Items, Vol. 01Документ800 страницU.S. ARMY - Encyclopedia of Explosives and Related Items, Vol. 01alexandraantonie1100% (3)
- The Encyclopedia of Explosives and Related Items PATR 2700 VOLUME 8Документ1 005 страницThe Encyclopedia of Explosives and Related Items PATR 2700 VOLUME 8Bloc22100% (3)
- Detonators, igniters, and primers guideДокумент22 страницыDetonators, igniters, and primers guidedjshyc100% (3)
- Nitro-Explosives: A Practical TreatiseОт EverandNitro-Explosives: A Practical TreatiseРейтинг: 2.5 из 5 звезд2.5/5 (3)
- NitrocelluloseДокумент7 страницNitrocellulosejumpupdnbdjОценок пока нет
- Acetone PeroxideДокумент2 страницыAcetone PeroxideRORTA.NET100% (15)
- The Encyclopedia of Explosives and Related Items PATR 2700 VOLUME 6Документ840 страницThe Encyclopedia of Explosives and Related Items PATR 2700 VOLUME 6Bloc22100% (2)
- Potassium Chlorate SynthesisДокумент3 страницыPotassium Chlorate SynthesisJohn RhodesОценок пока нет
- Thermite CompositionДокумент1 страницаThermite CompositionDexter LeeОценок пока нет
- Chlotares From Bleach and SaltДокумент16 страницChlotares From Bleach and SaltMaximiliano Muñoz García100% (1)
- AIChE Pocket HandbookДокумент64 страницыAIChE Pocket HandbookDinesh KanaujiyaОценок пока нет
- Complete Book of Flash PowderДокумент48 страницComplete Book of Flash PowderadamОценок пока нет
- CHE132 CPI REPORT ON EXPLOSIVES, PROPELLANTS AND TOXIC CHEMICAL AGENTSДокумент9 страницCHE132 CPI REPORT ON EXPLOSIVES, PROPELLANTS AND TOXIC CHEMICAL AGENTSEunice Flores100% (1)
- Joseph Abrusci - Professional Homemade Cherry BombsДокумент26 страницJoseph Abrusci - Professional Homemade Cherry BombsLê Nguyên ĐứcОценок пока нет
- PETN SynthesisДокумент4 страницыPETN SynthesisJack GraffОценок пока нет
- Triacetone Triperoxide (TATP)Документ1 страницаTriacetone Triperoxide (TATP)Beaune V. VillarazaОценок пока нет
- Non-Primary Explosive Detonator (NPED)Документ6 страницNon-Primary Explosive Detonator (NPED)partha das sharma100% (1)
- Purification of Laboratory Chemicals: Part 1 Physical Techniques, Chemical Techniques, Organic ChemicalsОт EverandPurification of Laboratory Chemicals: Part 1 Physical Techniques, Chemical Techniques, Organic ChemicalsРейтинг: 5 из 5 звезд5/5 (1)
- Chemistry and Technology of ExplosivesДокумент728 страницChemistry and Technology of ExplosivesFabian Borda100% (2)
- Preparatory Manual of Black Powder and PyrotechnicsДокумент334 страницыPreparatory Manual of Black Powder and PyrotechnicsDennis Danich83% (12)
- Nichrome Wire IgnitersДокумент3 страницыNichrome Wire IgnitersI wont write my name here :P100% (2)
- Chemistry of Powder and ExplosivesДокумент12 страницChemistry of Powder and Explosivesvnmaina100% (2)
- Acetone PeroxideДокумент3 страницыAcetone PeroxideI wont write my name here :P0% (1)
- Evil Detonator: Owner'S ManualДокумент5 страницEvil Detonator: Owner'S ManualMichael BrownОценок пока нет
- HMTDДокумент2 страницыHMTDIain Chilliemeister KingОценок пока нет
- Methods for Oxidation of Organic Compounds V2: Alcohols, Alcohol Derivatives, Alky Halides, Nitroalkanes, Alkyl Azides, Carbonyl Compounds Hydroxyarenes and AminoarenesОт EverandMethods for Oxidation of Organic Compounds V2: Alcohols, Alcohol Derivatives, Alky Halides, Nitroalkanes, Alkyl Azides, Carbonyl Compounds Hydroxyarenes and AminoarenesОценок пока нет
- TNT Trinitrotoluenes and Mono and DinitrotoluenesДокумент136 страницTNT Trinitrotoluenes and Mono and DinitrotoluenesWhiteOak ComenziОценок пока нет
- ASTM D 1356 - 00a Sampling and Analysis of AtmospheresДокумент14 страницASTM D 1356 - 00a Sampling and Analysis of Atmospheresalin2005Оценок пока нет
- Piric AcidДокумент2 страницыPiric Acidy_satyapОценок пока нет
- Nitro Compounds: Proceedings of the International Symposium Held at the Institute of Organic Synthesis, Polish Academy of Sciences, Warszawa, 18-20 September 1963От EverandNitro Compounds: Proceedings of the International Symposium Held at the Institute of Organic Synthesis, Polish Academy of Sciences, Warszawa, 18-20 September 1963Tadeusz UrbańskiРейтинг: 5 из 5 звезд5/5 (1)
- Synthesis of RDXДокумент4 страницыSynthesis of RDXmladen lakic100% (4)
- The Pyrotechnist's Treasury Or, Complete Art of Making FireworksОт EverandThe Pyrotechnist's Treasury Or, Complete Art of Making FireworksОценок пока нет
- 5) Nitroglycerin&DynamiteДокумент18 страниц5) Nitroglycerin&DynamiteMuhammad Irfan MalikОценок пока нет
- Science 9 exam questions on atomic structure and bondingДокумент3 страницыScience 9 exam questions on atomic structure and bondingLimar Anasco Escaso67% (3)
- Chemical reactions that release energy quicklyДокумент35 страницChemical reactions that release energy quicklyChetan SuranaОценок пока нет
- Making NitroglycerinДокумент4 страницыMaking Nitroglycerinmilad100% (1)
- Astm D971Документ4 страницыAstm D971JORGE SANTANDERОценок пока нет
- The Analysis of Explosives: Pergamon Series in Analytical ChemistryОт EverandThe Analysis of Explosives: Pergamon Series in Analytical ChemistryРейтинг: 5 из 5 звезд5/5 (2)
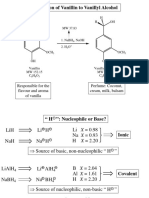
- Síntesis de Alcohol VainillílicoДокумент6 страницSíntesis de Alcohol VainillílicoYago L100% (1)
- 37 MM Launch Able MunitionsДокумент8 страниц37 MM Launch Able Munitionsali_winstonОценок пока нет
- Black Powder GradesДокумент2 страницыBlack Powder GradesPhileasОценок пока нет
- Explosive Plastic PHF-89 Technical DataДокумент2 страницыExplosive Plastic PHF-89 Technical DatahorscriОценок пока нет
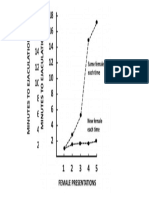
- Female Presentations: Same Female Each TimeДокумент1 страницаFemale Presentations: Same Female Each TimejoshuaericksonОценок пока нет
- Mustard StabilizationДокумент82 страницыMustard StabilizationjoshuaericksonОценок пока нет
- Political Activity and Public Demonstration Certificate of Completion PDFДокумент1 страницаPolitical Activity and Public Demonstration Certificate of Completion PDFjoshuaericksonОценок пока нет
- VX EvaporationДокумент7 страницVX EvaporationjoshuaericksonОценок пока нет
- Properties of Chemical Explosives and Explosive SimulantsДокумент334 страницыProperties of Chemical Explosives and Explosive Simulantsjoshuaerickson100% (2)
- Major Reaction Mechanism Decision Tree for SN1, SN2, E1 & E2 ReactionsДокумент1 страницаMajor Reaction Mechanism Decision Tree for SN1, SN2, E1 & E2 ReactionsjoshuaericksonОценок пока нет
- Precipitation Titrimetry AnalysisДокумент29 страницPrecipitation Titrimetry AnalysisPraveen KumarОценок пока нет
- (Junoon-E-Jee 3.0) Solid StateДокумент119 страниц(Junoon-E-Jee 3.0) Solid StateShiven DhaniaОценок пока нет
- Analytical Instruments in Water-Steam-Cycles: An Introduction For The Non-ChemistДокумент27 страницAnalytical Instruments in Water-Steam-Cycles: An Introduction For The Non-ChemistMaxi MaxiОценок пока нет
- Zero Export Steam Reforming Article - 1001307 PDFДокумент8 страницZero Export Steam Reforming Article - 1001307 PDFSakthi VelОценок пока нет
- Quantum Tutorial 2Документ2 страницыQuantum Tutorial 2Prathamesh KumarОценок пока нет
- Glancing Angle Deposition Method (GLAD)Документ13 страницGlancing Angle Deposition Method (GLAD)dhruv pratap singhОценок пока нет
- Ozone Layer & Greenhouse EffectДокумент29 страницOzone Layer & Greenhouse EffectIlyas A HuqqaniОценок пока нет
- 25) 8 ನೇ ತರಗತಿ ವಿಜ್ಞಾನ ಆಂಗ್ಲ ಮಾ.ಮಾ.ಪ್ರ.ಪ-1Документ9 страниц25) 8 ನೇ ತರಗತಿ ವಿಜ್ಞಾನ ಆಂಗ್ಲ ಮಾ.ಮಾ.ಪ್ರ.ಪ-1Abhisheck JatОценок пока нет
- Jurnal Symposium Lisat Agus Ismangil 2015Документ6 страницJurnal Symposium Lisat Agus Ismangil 2015a_ismangil_physicsОценок пока нет
- An Experimental Study On The Internal Corrosion of A Subsea Multiphase PipelineДокумент7 страницAn Experimental Study On The Internal Corrosion of A Subsea Multiphase PipelineYogaОценок пока нет
- CEX 5231 Ass 3 2015 - 2016Документ2 страницыCEX 5231 Ass 3 2015 - 2016MufeesОценок пока нет
- The "Golden Penny" DemonstrationДокумент3 страницыThe "Golden Penny" DemonstrationOren RosenfeldОценок пока нет
- Technical Proposal: Gypsum Powder Production Line With Capacity 60000 Tons Per YearДокумент25 страницTechnical Proposal: Gypsum Powder Production Line With Capacity 60000 Tons Per YearJohn TesfamariamОценок пока нет
- Determinación de 3 Alkil 2 Metoxipirazinas en Uvas Mostos y VinosДокумент9 страницDeterminación de 3 Alkil 2 Metoxipirazinas en Uvas Mostos y VinosEmmanuel BonninОценок пока нет
- Creep and FatigueДокумент20 страницCreep and Fatiguecheveresan123100% (2)
- Definition: Origin of ChargesДокумент5 страницDefinition: Origin of ChargesDavies MasumbaОценок пока нет
- 02 Assignments MECДокумент22 страницы02 Assignments MECWillis ChekovОценок пока нет
- Earth Science: Quarter 2 - Module 11Документ24 страницыEarth Science: Quarter 2 - Module 11dayna palaubsanonОценок пока нет
- Amines Amino Acids ProteinsДокумент13 страницAmines Amino Acids ProteinsClifford Dwight RicanorОценок пока нет
- High-Solids Epoxy Systems For Protective and Marine CoatingsДокумент6 страницHigh-Solids Epoxy Systems For Protective and Marine CoatingsJuan Carlos Contreras CherresОценок пока нет
- Page 1 of 3: What To Expect When Being Asked Boiling Point Questions On ExamsДокумент3 страницыPage 1 of 3: What To Expect When Being Asked Boiling Point Questions On ExamsSulochana KoviОценок пока нет
- EstañoДокумент646 страницEstañoJuan MartínezОценок пока нет
- B.Tech MOOCs Recommended Course ListДокумент41 страницаB.Tech MOOCs Recommended Course ListHarshit SrivastavaОценок пока нет
- Redox Titration Calculations and AnalysesДокумент7 страницRedox Titration Calculations and AnalysesAtikaRahayuОценок пока нет
- NSS Chemistry Part 15 Analytical Chemistry (Structural QuestionsДокумент42 страницыNSS Chemistry Part 15 Analytical Chemistry (Structural QuestionsKelvinNgОценок пока нет