Вам также может понравиться
- Optigal’s Q & A for the CLRE: Contact Lens Registry Exam Questions Basic Certification - NCLEОт EverandOptigal’s Q & A for the CLRE: Contact Lens Registry Exam Questions Basic Certification - NCLEОценок пока нет
- Theory and Technology of Multiscale Dispersed Particle Gel for In-Depth Profile ControlОт EverandTheory and Technology of Multiscale Dispersed Particle Gel for In-Depth Profile ControlОценок пока нет
- TR0051 Elute From PolyacrylamideДокумент2 страницыTR0051 Elute From PolyacrylamideKasagalaОценок пока нет
- Protocolo Purificacion Desde GelДокумент3 страницыProtocolo Purificacion Desde GelAriel ArayaОценок пока нет
- Diamond Nucleic Acid Dye: Technical ManualДокумент8 страницDiamond Nucleic Acid Dye: Technical Manualamelia nurdiniОценок пока нет
- Lonza BenchGuides SourceBook Section VI - Recovery of DNA From Agarose GelsДокумент10 страницLonza BenchGuides SourceBook Section VI - Recovery of DNA From Agarose GelsVijaya SubramaniОценок пока нет
- Protocolo Gelred PDFДокумент2 страницыProtocolo Gelred PDFRodrigo UchyamaОценок пока нет
- GelCode Blue Stain ReagentДокумент4 страницыGelCode Blue Stain ReagentAhammed Abu DilОценок пока нет
- DGGEДокумент7 страницDGGENupur NagavekarОценок пока нет
- Diamond Nucleic Acid Dye FB142Документ1 страницаDiamond Nucleic Acid Dye FB142Stephen SabinayОценок пока нет
- Bulletin 6201Документ4 страницыBulletin 6201nia lutfianaОценок пока нет
- Promega Wizard SV Gel and PCR Clean Up SystemДокумент13 страницPromega Wizard SV Gel and PCR Clean Up Systemandrea nettle bareaОценок пока нет
- SDS-Polyacrylamide Gel Electrophoresis of ProteinsДокумент9 страницSDS-Polyacrylamide Gel Electrophoresis of ProteinsYoung LoveОценок пока нет
- litdoc71717700AL 2011083021560Документ28 страницlitdoc71717700AL 2011083021560LilyОценок пока нет
- 7 Autoradiograms 35S and 32P - 1987 - Methods in EnzymologyДокумент7 страниц7 Autoradiograms 35S and 32P - 1987 - Methods in EnzymologyMontsZs G-oОценок пока нет
- Gel PrepДокумент2 страницыGel PrepRijjy On FreedayОценок пока нет
- Autotype Indirect Photostencil Films GuideДокумент4 страницыAutotype Indirect Photostencil Films GuideTitus MendezОценок пока нет
- Lonza Bench Guides Source Book Section XII - Iso Electric Focusing of Proteins On Agarose GelsДокумент18 страницLonza Bench Guides Source Book Section XII - Iso Electric Focusing of Proteins On Agarose GelsozlemunaldiОценок пока нет
- Resolving 25-700bp DNA Fragments with Acrylamide GelsДокумент4 страницыResolving 25-700bp DNA Fragments with Acrylamide GelsNoviana WulansariОценок пока нет
- Jobsheet 2 (Protein Demo)Документ7 страницJobsheet 2 (Protein Demo)nurul izzah bahtiarОценок пока нет
- Data Sheet: HDSP SeriesДокумент10 страницData Sheet: HDSP SeriesFirmansyahPutraОценок пока нет
- pGEM VectorДокумент10 страницpGEM VectorAnil KumarОценок пока нет
- Trackit 1kbplus ManДокумент4 страницыTrackit 1kbplus Manthumita kumiОценок пока нет
- Technical Bulletin: Proteosilver Silver Stain KitДокумент4 страницыTechnical Bulletin: Proteosilver Silver Stain KitAjeng FadillahОценок пока нет
- MW MarkorДокумент4 страницыMW MarkorEdonОценок пока нет
- Lab 4.isolation of PlasmidДокумент7 страницLab 4.isolation of PlasmidJane MargarethaОценок пока нет
- Agarose Gel Electrophoresis DNAДокумент5 страницAgarose Gel Electrophoresis DNAAli Hasan100% (1)
- Exp 7-SDS-PAGEДокумент18 страницExp 7-SDS-PAGERadwan M SaadehОценок пока нет
- Fast BlastДокумент14 страницFast BlastManuelSBОценок пока нет
- DNA-RNA-protein Analyze Part 1,2 2016Документ33 страницыDNA-RNA-protein Analyze Part 1,2 2016Meliana LiuОценок пока нет
- GeneJET Gel Extraction KitДокумент10 страницGeneJET Gel Extraction KitJocelyn OrellanaОценок пока нет
- Sds-Polyacrylamide Gel Electrophoresis IntroductionДокумент5 страницSds-Polyacrylamide Gel Electrophoresis IntroductionmejohОценок пока нет
- Gel Filtration ChromatographyДокумент3 страницыGel Filtration ChromatographyFadli ArchieОценок пока нет
- CEQChemistryProtocol Dye Terminator SequencerДокумент20 страницCEQChemistryProtocol Dye Terminator SequencerAhmad SalehОценок пока нет
- A1-Specimen Preparation ProtocolДокумент0 страницA1-Specimen Preparation ProtocolidownloadbooksforstuОценок пока нет
- ASTM D 3156 - 96 (Reapproved 2001) Rubber-Chromatographic Analysis of AntidegradantsДокумент5 страницASTM D 3156 - 96 (Reapproved 2001) Rubber-Chromatographic Analysis of Antidegradantsalin2005Оценок пока нет
- Complete Teaching Kit For PCR Amplification: 25 Rxns of 50 L OR 50 Rxns of 25 LДокумент5 страницComplete Teaching Kit For PCR Amplification: 25 Rxns of 50 L OR 50 Rxns of 25 LSmitha KollerahithluОценок пока нет
- Quantam Freeze and Squeeze ProtocolДокумент1 страницаQuantam Freeze and Squeeze Protocolme_dayakarОценок пока нет
- CDNA ExtractionДокумент4 страницыCDNA Extractionvikashisar009Оценок пока нет
- Phusion Taq Manuale0553Документ16 страницPhusion Taq Manuale0553César UsléОценок пока нет
- DGGE Analysis of Symbiodinium ITS2 for Strain DifferentiationДокумент9 страницDGGE Analysis of Symbiodinium ITS2 for Strain Differentiationsathish.botОценок пока нет
- TDS-JOTACOTE 5-EnglishДокумент4 страницыTDS-JOTACOTE 5-EnglishthirdОценок пока нет
- Purelink Quick Gel Extraction Kit ManДокумент24 страницыPurelink Quick Gel Extraction Kit ManNidia ALice PinheiroОценок пока нет
- Protocolo Gelred BiotiumДокумент4 страницыProtocolo Gelred BiotiummaryОценок пока нет
- Virtual Drug Screening Stream Spring 2011: Lab: Protein CharacterizationДокумент5 страницVirtual Drug Screening Stream Spring 2011: Lab: Protein CharacterizationKeri Gobin SamarooОценок пока нет
- Hardness, Total Ultra Low Range - AP-67 - 1900Документ3 страницыHardness, Total Ultra Low Range - AP-67 - 1900Rizka MaharanaОценок пока нет
- SDS-PAGE Guide: Separate Proteins by Size & ChargeДокумент39 страницSDS-PAGE Guide: Separate Proteins by Size & ChargeSnehalphirke100% (1)
- Jahirul IslamДокумент20 страницJahirul IslamPronoy 01Оценок пока нет
- Photostencil Emulsions 23Документ4 страницыPhotostencil Emulsions 23huroncursiОценок пока нет
- Guide To Manual Processing of NDT Films: Technical Data / Non-Destructive TestingДокумент6 страницGuide To Manual Processing of NDT Films: Technical Data / Non-Destructive TestingcarlosОценок пока нет
- Western Blot ProtocolДокумент35 страницWestern Blot ProtocolH DegaïchiaОценок пока нет
- Millipore UF Membranes Protocol (Ultracel) PDFДокумент2 страницыMillipore UF Membranes Protocol (Ultracel) PDFR YОценок пока нет
- Devthane 379H: Aliphatic Urethane Gloss EnamelДокумент2 страницыDevthane 379H: Aliphatic Urethane Gloss EnamelBikeBook EnsenadaОценок пока нет
- Genematrix Universal Dna/Rna/Protein Purification KitДокумент16 страницGenematrix Universal Dna/Rna/Protein Purification Kitsatnam thindОценок пока нет
- MCQs IPДокумент45 страницMCQs IPJayendrasingh Bayas93% (14)
- GoTaq Green Master MixДокумент2 страницыGoTaq Green Master MixDody GogaОценок пока нет
- Jotacote 5Документ4 страницыJotacote 5Ridwan BaharumОценок пока нет
- Datasheet JotunДокумент12 страницDatasheet JotunbayuОценок пока нет
- SDS PAGE de ProteinasДокумент11 страницSDS PAGE de Proteinasana cristinaОценок пока нет
- Medicinal PlantДокумент11 страницMedicinal Plantpati_deeps234Оценок пока нет
- 2012A High Throughput DNA Extraction MethodДокумент7 страниц2012A High Throughput DNA Extraction MethodOsama AbdulkareemОценок пока нет
- 2009radiation Inactivation of Galactose PDFДокумент6 страниц2009radiation Inactivation of Galactose PDFOsama AbdulkareemОценок пока нет
- 2011direct Effects of Ionizing Radiation On Macromolecules PDFДокумент8 страниц2011direct Effects of Ionizing Radiation On Macromolecules PDFOsama AbdulkareemОценок пока нет
- 11handbook of Detection of Enzyme On Electrophoret GelsДокумент570 страниц11handbook of Detection of Enzyme On Electrophoret GelsOsama Abdulkareem100% (2)
- Journal of Cereal Science - v50 - Iss2Документ170 страницJournal of Cereal Science - v50 - Iss2Osama AbdulkareemОценок пока нет
- 2002effect of Radiation On Plant Cells PDFДокумент12 страниц2002effect of Radiation On Plant Cells PDFOsama AbdulkareemОценок пока нет
- Journal of Cereal Science - v49 - Iss1Документ166 страницJournal of Cereal Science - v49 - Iss1Osama AbdulkareemОценок пока нет
- Chap 12 Biotechnology and Its ApplicationsДокумент3 страницыChap 12 Biotechnology and Its ApplicationsRajarshiОценок пока нет
- Cell Cycle StagesДокумент16 страницCell Cycle StagesSean0% (1)
- Function of PhotosynthesisДокумент58 страницFunction of PhotosynthesisJulian ChristopherОценок пока нет
- pGL2 Luciferase Reporter Vectors ProtocolДокумент26 страницpGL2 Luciferase Reporter Vectors ProtocolChen GaoОценок пока нет
- 2020 03 31 016972v2 FullДокумент123 страницы2020 03 31 016972v2 FullEmma LouthОценок пока нет
- UT Dallas Syllabus For hcs7372.001.11s Taught by Jonathan Ploski (Jep101000)Документ5 страницUT Dallas Syllabus For hcs7372.001.11s Taught by Jonathan Ploski (Jep101000)UT Dallas Provost's Technology GroupОценок пока нет
- AviChem RXG Brochure - Viewing FileДокумент4 страницыAviChem RXG Brochure - Viewing FileAhmad GhОценок пока нет
- Role of Oxidative Stress in Biological Systems: December 2019Документ9 страницRole of Oxidative Stress in Biological Systems: December 2019Ahmad AliОценок пока нет
- Ap Cell Division and Reproduction MCДокумент6 страницAp Cell Division and Reproduction MCapi-522349089Оценок пока нет
- Illumina Genome Analyzer Maintenance GuideДокумент66 страницIllumina Genome Analyzer Maintenance GuideTito DANGER Jankowski0% (1)
- Wednesday 20 January 2021: BiologyДокумент40 страницWednesday 20 January 2021: BiologyMilka RahmanОценок пока нет
- Pharmacology AT GLIMPSE PDFДокумент252 страницыPharmacology AT GLIMPSE PDFBiswarup Das100% (1)
- Pharmaceutical Biotechnology PDFДокумент24 страницыPharmaceutical Biotechnology PDFShyamlaОценок пока нет
- Regulation of the Cell Cycle by Protein KinasesДокумент5 страницRegulation of the Cell Cycle by Protein KinasesmohsinОценок пока нет
- Drugs in The Pipeline For HBVДокумент21 страницаDrugs in The Pipeline For HBVMarius StancuОценок пока нет
- The parts and functions of cellsДокумент7 страницThe parts and functions of cellsKate Aireen JerezОценок пока нет
- Genetic MutationДокумент27 страницGenetic MutationKathleen JOy BiasUraОценок пока нет
- Microbiology Jeopardy ReviewДокумент31 страницаMicrobiology Jeopardy Reviewjmiller623Оценок пока нет
- The Moss Physcomitrium (Physcomitrella) Patens: A Model Organism For Non-Seed PlantsДокумент16 страницThe Moss Physcomitrium (Physcomitrella) Patens: A Model Organism For Non-Seed PlantsRski AmelОценок пока нет
- 8.1 and 8.2 Group AssignmentДокумент7 страниц8.1 and 8.2 Group AssignmentDaniel CondreayОценок пока нет
- Protein Purification Poster ProДокумент1 страницаProtein Purification Poster ProIsaac Nicholas NotorioОценок пока нет
- Farmacos y ConclusionДокумент1 страницаFarmacos y Conclusionkarla torrezОценок пока нет
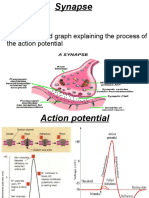
- Synapse Structure and Function ExplainedДокумент11 страницSynapse Structure and Function ExplainedMuhammad AbdullahОценок пока нет
- Cell BiologyДокумент8 страницCell BiologyRichard JomeОценок пока нет
- Protein Structure Powerpoint Presentation Newest Pink and PurpleДокумент21 страницаProtein Structure Powerpoint Presentation Newest Pink and Purpleapi-281150432Оценок пока нет
- Car-T Cell 2 (7757)Документ8 страницCar-T Cell 2 (7757)Saradha PellatiОценок пока нет
- Flow Cytometry Capabilities GuideДокумент16 страницFlow Cytometry Capabilities GuideirmaОценок пока нет
- AP1 AP2 PCR Method For AHPND Detection - Flegel 2013Документ6 страницAP1 AP2 PCR Method For AHPND Detection - Flegel 2013Alden ReynaОценок пока нет
- DNA Restriction and Ligation in Gene Cloning, A Study Case in Prolidase Gene Cloning From Lactic Acid BacteriaДокумент11 страницDNA Restriction and Ligation in Gene Cloning, A Study Case in Prolidase Gene Cloning From Lactic Acid BacteriaRina ZaniaОценок пока нет
- MMBIO Exam1 Review - 241Документ23 страницыMMBIO Exam1 Review - 241Riley SarabiaОценок пока нет