6.
Phase Diagrams
Gibbs Phase Rule
Formal denition of phase: A phase is a state of mat-
ter that is uniform throughout, not only in chemical
composition but also in physical state.
Denition of number of components: The number of
components is the minimum number of independent
species necessary to dene the composition of all the
phases in the system.
Equivalent denition: The number of components is
the number of distinct chemical species in the system
less the number of constraints on the concentration of
these species.
Constraints: Constraints are conditions that gives
rise to additional relations between the chemical po-
tentials or between intensive variables, for instance,
mole fractions in a given phase. Typical examples
are: (i) Each independent chemical reaction provides
one relation between the chemical potentials, namely
PChem I 6.1
the equilibrium condition
i
i
= 0 (see next Chap-
ter). (The
i
s are the stoichiometric coefcients of
the reacting species, positive for products and nega-
tive for reactants.) Chemical reactions are indepen-
dent if no reaction can be written as a combination
of the others. (ii) Material balance or stoichiometric
relations. (iii) Electroneutrality condition (for ionic so-
lutions).
Degrees of freedom: The number of independent
intensive variables needed to describe a system is
called its degrees of freedom or variance.
Since the chemical potential is an intensive quanti-
ty, we can use n
t ot
= 1 to evaluate its value, and we
have
(T, P, n
1
, . . . , n
N
) =(T, P, x
1
, . . . , x
N
)
i.e., we can characterize the composition of the sys-
tem, or a given phase, by the set of mole fractions
instead of the set of numbers of moles. Further, since
N
i
x
i
= 1, we need only N 1 mole fractions, say
{x
1
, x
2
, . . . , x
N1
}.
PChem I 6.2
Consider a system with C components and phas-
es. (We use , instead of the textbooks P, for the
number of phases to avoid confusion with the pres-
sure variable.) To specify the state of such a system,
we need to know the intensive variables
T, P, {x
(1)
1
, x
(1)
2
, . . . , x
(1)
C1
}, {x
(2)
1
, x
(2)
2
, . . . , x
(2)
C1
},
. . . , {x
()
1
, x
()
2
, . . . , x
()
C1
}
namely 2+(C 1) variables.
On the other hand, at equilibrium the chemical po-
tential of species J must be the same throughout the
system:
(1)
J
=
(2)
J
= =
()
J
for J = 1, . . . ,C. In other words, there are 1 equa-
tions for each component, and there are C compo-
nents, so that we have C(1) equations overall that
establish relations between the 2+(C1) variables.
Thus the degrees of freedom are
F =2+(C 1) C (1)
PChem I 6.3
F =2+C
This is the Gibbs phase rule.
Practice:
How many components are in the following systems:
(a) water, allowing for its ionization
(b) AlCl
3
in water, noting that hydrolysis and precipi-
tation of Al(OH)
3
occur.
In the following, let S denote the number of distinct
chemical species, and R the number of constraints.
(a) The species are H
2
O, H
+
, OH
, i.e., S = 3. The
constraints are:
equilibrium:
H
2
O
H
+
+OH
electroneutrality:
n
H
+
(aq) =n
OH
(aq)
PChem I 6.4
divide by n
tot
(aq)
x
H
+
(aq) =x
OH
(aq)
So, R =2 and C =S R =1
(b) AlCl
3
, Al
3+
, Cl
, Al(OH)
3
, H
2
O, H
+
, OH
so, S =7
equilibria:
AlCl
3
(s)
Al
3+
(aq) +3Cl
(aq)
H
2
O(l)
H
+
(aq) +OH
(aq)
Al
3+
(aq) +3OH
(aq)
Al(OH)
3
(s)
electroneutrality:
n
H
+
(aq) +3n
Al
3+
(aq) =n
OH
(aq) +n
Cl
(aq)
divide by n
tot
(aq)
x
H
+
(aq) +3x
Al
3+
(aq) =x
OH
(aq) +x
Cl
(aq)
PChem I 6.5
So R =3+1 and C =S R =74 =3
one-component systems
F =C +2 =3
3
=1 : F =2 P and T can be freely chosen: area
=2 : F =1 P =P(T) or T =T(P) phase coexistence line
=3 : F =0 P and T are xed; triple point
[Figure: one-component phase diagram, Fig. 6.2]
locating rst-order phase transition points: thermal
analysis, cooling curve
[Figure: cooling curve, Fig. 6.4]
two-component systems
F =4
4
PChem I 6.6
at most 4 phases can coexist; this occurs at a point in
a T-P-x-diagram
vapor pressure diagrams for mixture of volatile liq-
uids
liquid-vapor equilibriumof a mixture of similar liquids,
e.g., toluene and benzene: ideal solution
P
A
=x
A
P
A
, P
B
=x
B
P
B
x
i
= mole fraction of i in the liquid phase
P =P
A
+P
B
=x
A
P
A
+x
B
P
B
=x
A
P
A
+(1x
A
)P
B
P =P
B
+
_
P
A
P
B
_
x
A
total vapor pressure at xed temperature varies lin-
early with the composition of the liquid phase
PChem I 6.7
phase di-
agram
assumes
A is more
volatile, i.e.,
P
A
>P
B
since A is more volatile, vapor will be richer in A: com-
position of liquid phase and vapor phase in equilibri-
um are not the same
mole fractions in the vapor phase: y
A
, y
B
y
A
=
P
A
P
, y
B
=
P
B
P
y
A
=
x
A
P
A
P
B
+
_
P
A
P
B
_
x
A
=
x
A
P
B
P
A
+
_
1
P
B
P
A
_
x
A
y
A
=
x
A
x
A
+
P
B
P
A
x
B
>x
A
for P
A
>P
B
PChem I 6.8
x
A
=
y
A
P
B
P
_
P
A
P
B
_
y
A
P =
P
A
P
B
P
A
+
_
P
B
P
A
_
y
A
to understand distillation, use a phase diagram that
contains both the liquid phase and the vapor phase
composition
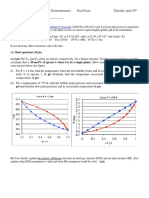
[Figure: vapor pressure phase diagrams, Fig. 6.9, 6.10, & 6.12]
vertical dotted line = isopleth
horizontal lines a
1
-a
1
, etc., = tie lines
relative amounts of liquid and vapor phase: lever rule
n
=n
[Figure: lever rule, Fig. 6.13]
temperature-composition diagrams
distillation of mixtures
PChem I 6.9
[Figure: distillation, Fig. 6.14]
[Figure: fractional distillation, Fig. 6.15]
deviations from ideal solutions
mixing, real solutions
real solutions:
AB
=
AA
and
AB
=
BB
;
i j
= average
interaction energy between particles i and j
G
i
=n
A
A
(l ) +n
B
B
(l )
G
f
=n
A
_
A
(l ) +RT lna
A
_
+
+n
B
_
B
(l ) +RT lna
B
_
mix
G =nRT
_
x
A
lna
A
+x
B
lna
B
_
mix
G =RT
_
x
A
lna
A
+x
B
lna
B
_
a
i
=
i
x
i
mix
G =RT
_
x
A
lnx
A
+x
A
ln
A
+x
B
lnx
B
+x
B
ln
B
_
mix
G =RT
_
x
A
lnx
A
+x
B
lnx
B
_
+
+RT
_
x
A
ln
A
+x
B
ln
B
_
mix
G =
mix
G
ideal
+G
E
G
E
=RT
_
x
A
ln
A
+x
B
ln
B
_
PChem I 6.10
If
AB
>
AA
and
AB
>
BB
, then the A and B molecules
decrease each others escaping tendency. Favorable
interactions between A and B molecules reduce the
vapor pressure of the mixture below the ideal value,
i.e., the AB interactions stabilize the liquid phase =
negative deviations from Raoults law, <1 and G
E
<
0, more favorable to mixing than ideal.
example: acetone and chloroform can form hydro-
gen bonds, a relatively strong intermolecular force
not present in the pure liquids:
[Figure: vapor pressure acetone/chloroform mixture]
negative deviations from ideality may lead to a maxi-
mum in the temperature-composition diagram
[Figure: high-boiling azeotrope, Fig. 6.16]
repeated distillation of the liquid leads to an
azeotrope: evaporation occurs without change in
composition
PChem I 6.11
example: hydrochloric acid/water, azeotropic at 80%
by mass of water, boils unchanged at 108.6
C
If
AB
<
AA
, then the solute breaks up strong inter-
molecular forces in the solvent (nearly always in-
volves molecules containing OH groupswater, al-
cohols, organic acidswhich may form hydrogen
bonds). Unfavorable interactions between A and B
molecules increase the volatility of the liquids, i.e.,
the AB interactions destabilize the liquid phase =
positive deviations from Raoults law, > 1 and G
E
>
0, less favorable to mixing than ideal.
example ethanol/heptane
[Figure: vapor pressure ethanol/heptane mixture]
[Figure: activity of ethanol in heptane]
positive deviations from ideality may lead to a mini-
mum in the temperature-composition diagram
[Figure: low-boiling azeotrope, Fig. 6.17]
fractional distillation of the vapor leads to an
azeotrope
example: ethanol/water, azeotropic at 4% by mass of
water, boils unchanged at 78
C
PChem I 6.12
liquid-liquid phase diagrams: partially miscible liquids
[Figure: phase diagram of hexane/nitrobenzene, Fig. 6.19 & 6.20]
upper critical solution temperature or upper conso-
lute temperature T
uc
can be modeled by regular solutions
molecules mix randomly as they do in ideal solutions:
S
E
=0
deviations from ideality due to different strength of
interactions between like and unlike molecules: H
E
=
0
must have H
E
0 as x
A
0 or x
B
0
simplest form that fullls this condition:
H
E
=wx
A
x
B
=RTx
A
x
B
with =w/RT
H
E
symmetric in x
A
and x
B
=1x
A
[Figure: H
E
/nRT vs x
A
, Fig. 5.19]
PChem I 6.13
w
AA
+
BB
2
AB
w is generally weakly temperature and composition
dependent; will be neglected (otherwise we do not
have strictly speaking a regular solution)
G
E
=wx
A
x
B
=RTx
A
x
B
V
E
=
_
_
G
E
P
_
_
T
=0
U
E
=wx
A
x
B
the activity coefcients for regular solutions are given
by the Margules equations:
A
=exp
_
wx
2
B
RT
_
,
B
=exp
_
wx
2
A
RT
_
and thus
a
A
=
A
x
A
=x
A
exp
_
w(1x
A
)
2
RT
_
PChem I 6.14
P
A
=
_
x
A
exp
_
w(1x
A
)
2
RT
__
P
A
[Figure: vapor pressure of solvent for a regular solution, Fig. 5.32]
if w < 0, interactions are stronger between unlike
molecule than like molecules: negative deviation
from Raoults law; more favorable mixing; mixing is
exothermic:
mix
H = H
E
<0
in fact,
mix
G < 0, i.e., mixing is spontaneous for all
compositions,
mix
G has one deep minimum at x
A
=
1/2 =
mix
G
x
2
A
0 for all x
A
=mixing is homogeneous, no phase separation oc-
curs
if w >0, interactions are stronger between like mole-
cule than unlike molecules: positive deviation from
Raoults law; less favorable mixing; mixing is en-
dothermic:
mix
H = H
E
>0;
PChem I 6.15
since
mix
G
ideal
= RT
_
x
A
lnx
A
+x
B
lnx
B
_
0 and G
E
=
wx
A
x
B
0,
mix
G may have a maximum at x
A
= 1/2
and two minima x
I ,I I
A
located symmetrically on either
side of 1/2, i.e.,
mix
G
x
2
A
<0 for a range of x
A
[Figure: Gibbs free energy of mixing for regular solutions, Fig. 5.20]
=phase separation occurs; mixture of composition
x
A
with x
I
A
< x
A
< x
I I
A
separates into phases with com-
position x
I
A
and x
I I
A
, respectively; the proportions of
the two phases are given by the lever rule
since
mix
G
ideal
T and G
E
is temperature indepen-
dent,
mix
G is expected to develop a maximum and
two minima if the interaction term, G
E
, becomes im-
portant enough compared to the ideal mixing term,
mix
G
ideal
, i.e., if T is sufciently small
more precisely, if w >0, then for T <T
uc
(upper con-
solute temperature),
mix
G has two minima, corre-
sponding to two phases, one rich in A and the other
rich in B
PChem I 6.16
if T >T
uc
, then
mix
G has only one minimum
at T = T
uc
,
mix
G has a degenerate or double mini-
mum; this corresponds to a critical point of the sys-
tem
critical point: rst three derivatives of the Gibbs en-
ergy vanish simultaneously, the fourth is positive
determine the upper consolute temperature for a reg-
ular solution
mix
G =RT
_
x
A
lnx
A
+x
B
lnx
B
_
+wx
A
x
B
mix
G =RT
_
x
A
lnx
A
+(1x
A
) ln(1x
A
) +
+x
A
(1x
A
)
_
mix
G
x
A
=RT
_
1 lnx
A
+x
A
1
x
A
+
+(1) ln(1x
A
) +(1x
A
)
1
(1x
A
)
(1)+
+(12x
A
)
_
=0
PChem I 6.17
RT
_
ln
_
x
A
1x
A
_
+(12x
A
)
_
=0
mix
G
x
2
A
=RT
_
1
x
A
+
1
1x
A
2
_
=0
mix
G
x
3
A
=RT
_
1
x
2
A
1
(1x
A
)
2
(1)
_
=0
= x
A,c
=
1
2
2nd derivative = 0 =
c
=2 =
w
RT
c
=T
c
=
w
2R
rst derivative vanishes at x
A
= 1/2 due to the sym-
metry of the model, and it is easily veried that
the fourth derivative is positive at the critical point
(x
A,c
, T
c
)
[Figure: location of phase boundary, Fig. 6.23]
mixture of components that form weak complexes at
low temperatures may have a lower critical solution
PChem I 6.18
temperature or lower consolute temperature T
l c
[Figure: water/triethylamine mixture, Fig. 6.24]
some systems have both upper and lower consolute
temperatures
[Figure: water/nicotine mixture, Fig. 6.25]
PChem I 6.19
Вам также может понравиться
- Lecture-8,9,10 VLE DiagramsДокумент64 страницыLecture-8,9,10 VLE DiagramsShiavm PatelОценок пока нет
- CBE3508 Sp21 FinalДокумент6 страницCBE3508 Sp21 Finalsasuke uchihaОценок пока нет
- Vapor Liquid Equilibria: A Review: Maya B. Mane and S. N. ShindeДокумент15 страницVapor Liquid Equilibria: A Review: Maya B. Mane and S. N. ShindeDesi Riana SaputriОценок пока нет
- 2008-2D-RCM-methyl AcetateДокумент13 страниц2008-2D-RCM-methyl Acetatemaissam ferdosiОценок пока нет
- A Self-Consistent GE MR For CEoS Derivation and Fugacity CoefficientsДокумент4 страницыA Self-Consistent GE MR For CEoS Derivation and Fugacity Coefficientsmurdanetap957Оценок пока нет
- The Gibbs Phase Rule RevisitedДокумент3 страницыThe Gibbs Phase Rule Revisitedleizar_death640% (1)
- Azeotropic DiagramДокумент13 страницAzeotropic DiagramamoОценок пока нет
- VLE: Vapor Liquid EquilibriumДокумент39 страницVLE: Vapor Liquid EquilibriumTouhid IslamОценок пока нет
- Activity Coefficient Models Describe EVL Binay Systems Ionic LiquidsДокумент14 страницActivity Coefficient Models Describe EVL Binay Systems Ionic LiquidsJulian VargasОценок пока нет
- Phases and Solutions Phase Diagram GuideДокумент66 страницPhases and Solutions Phase Diagram GuideSYUHADAFAATAHОценок пока нет
- 1-Vle Part 1Документ30 страниц1-Vle Part 1Arfa Zulkifli01Оценок пока нет
- Binous Nasri PREOS MatlabДокумент10 страницBinous Nasri PREOS MatlabLuis Carlos CabreraОценок пока нет
- 06 Atkins Chap06Документ16 страниц06 Atkins Chap06tatianarafaОценок пока нет
- Phase RuleДокумент9 страницPhase RuleMadhavanIceОценок пока нет
- Chapter 6 - Multiphase Systems: CBE2124, LevickyДокумент27 страницChapter 6 - Multiphase Systems: CBE2124, LevickyRimmonОценок пока нет
- Module V Phase & Chem EqbДокумент26 страницModule V Phase & Chem Eqbarhanbhandawat66Оценок пока нет
- Solution ThermoДокумент9 страницSolution ThermofarahanisiliasОценок пока нет
- Experiment 2Документ3 страницыExperiment 2grj_076Оценок пока нет
- Phase DiagramsДокумент19 страницPhase Diagramsget2csОценок пока нет
- Vle For DummiesДокумент8 страницVle For Dummiesira_rancicОценок пока нет
- 254 8 Liquid Vapour EquilibriumДокумент6 страниц254 8 Liquid Vapour EquilibriumJustina JankauskaitėОценок пока нет
- Distillation Lec 2Документ12 страницDistillation Lec 2Omer IbrahimОценок пока нет
- Phase Equilibria Two-Component System: I. Liquid-Liquid System Ideal SolutionДокумент16 страницPhase Equilibria Two-Component System: I. Liquid-Liquid System Ideal SolutionChelsea MartinezОценок пока нет
- Chemical ReactorДокумент49 страницChemical ReactorAjay SatputeОценок пока нет
- CL-201 Chapter 6 Multiphase Systems (Compatibility Mode) PDFДокумент42 страницыCL-201 Chapter 6 Multiphase Systems (Compatibility Mode) PDFSuman MandalОценок пока нет
- Phase EquilibriaДокумент14 страницPhase EquilibriaPaden TranОценок пока нет
- Fe Chemical EngineeringДокумент5 страницFe Chemical EngineeringJudith LugoОценок пока нет
- Thermodynamics Separations BasicsДокумент12 страницThermodynamics Separations BasicsSata AjjamОценок пока нет
- Atkins-chapter06.lect01Документ43 страницыAtkins-chapter06.lect01雅萍 俞Оценок пока нет
- Improvement of The Van Der Waals Equation of StateДокумент13 страницImprovement of The Van Der Waals Equation of StateRené Mora-CasalОценок пока нет
- Equilibrium Stage Processes Flash VaporizationДокумент30 страницEquilibrium Stage Processes Flash Vaporizationj0haNN3sОценок пока нет
- Computing Liquid-Vapor Phase Diagrams For Non-Ideal Binary MixturesДокумент22 страницыComputing Liquid-Vapor Phase Diagrams For Non-Ideal Binary Mixturesmurdanetap957Оценок пока нет
- Vapor Liquid EquilibriumДокумент28 страницVapor Liquid EquilibriumKhloud MadihОценок пока нет
- SY Second Term, Phase RuleДокумент20 страницSY Second Term, Phase RuleBapu ThoratОценок пока нет
- Distillation - Dr.K.Suresh - NotesДокумент63 страницыDistillation - Dr.K.Suresh - NotesRoyalОценок пока нет
- Chemical Examples For The Fit Equations: ExampleДокумент11 страницChemical Examples For The Fit Equations: Exampleskrim240Оценок пока нет
- Liq Vap Phase 2Документ11 страницLiq Vap Phase 2Xedice MemmedyarovaОценок пока нет
- AlgoДокумент46 страницAlgoJoseCastilhoОценок пока нет
- Multiphase Systems ExplainedДокумент53 страницыMultiphase Systems Explainedعراقية KHОценок пока нет
- Chem RXN EquilДокумент12 страницChem RXN EquilfarahanisiliasОценок пока нет
- Phase Diagrams: Example, Draw MacrostructureДокумент54 страницыPhase Diagrams: Example, Draw MacrostructureMilan NaikОценок пока нет
- 71 - Galicia1986Документ9 страниц71 - Galicia1986Moltimer Folchart CrawОценок пока нет
- Two ComponentsДокумент19 страницTwo ComponentsMumtaz AhmadОценок пока нет
- 195 Sample-ChapterДокумент12 страниц195 Sample-ChapterGently Genius GenieОценок пока нет
- ReportДокумент4 страницыReportChadt Montague I'gautteОценок пока нет
- Separation Process Engineering CHEN 312: Ys18@aub - Edu.lbДокумент28 страницSeparation Process Engineering CHEN 312: Ys18@aub - Edu.lbsoe0303Оценок пока нет
- Single Equilibrium Stages and Flash CalculationsДокумент20 страницSingle Equilibrium Stages and Flash CalculationsMohd ImranОценок пока нет
- Chapter 5 - Nahid - July 2017Документ32 страницыChapter 5 - Nahid - July 2017Abdul BariОценок пока нет
- Phase RuleДокумент27 страницPhase RulejaiminОценок пока нет
- Michelsen The Isothermal Flash Problem 2 PhasДокумент20 страницMichelsen The Isothermal Flash Problem 2 Phasgggggg82Оценок пока нет
- 0378 38122987010 7Документ15 страниц0378 38122987010 7Tiên PhạmОценок пока нет
- Phase 3Документ35 страницPhase 3Faria Sultana MimiОценок пока нет
- Distillation TheoryДокумент40 страницDistillation TheoryIrvin HernandezОценок пока нет
- Calculation of Phase Envelopes and Critical Points For Multicomponent MixturesДокумент10 страницCalculation of Phase Envelopes and Critical Points For Multicomponent Mixturesflavio_cordero_1Оценок пока нет
- Unit 7 PhaseruleДокумент11 страницUnit 7 Phaseruleengineeringchemistry100% (2)
- Volume Additivity 1Документ14 страницVolume Additivity 1Kenneth Mendoza SorianoОценок пока нет
- Working Guide to Vapor-Liquid Phase Equilibria CalculationsОт EverandWorking Guide to Vapor-Liquid Phase Equilibria CalculationsРейтинг: 5 из 5 звезд5/5 (1)
- Reactive Oxygen Species: Signaling Between Hierarchical Levels in PlantsОт EverandReactive Oxygen Species: Signaling Between Hierarchical Levels in PlantsFranz-Josef SchmittОценок пока нет
- KVBA Hive Inspection SheetДокумент1 страницаKVBA Hive Inspection SheetKaaya GodfreyОценок пока нет
- 2021 - MCH 7105 - Advances in ElectrochemistryДокумент32 страницы2021 - MCH 7105 - Advances in ElectrochemistryKaaya GodfreyОценок пока нет
- List of Certified Seedling Nurseries 2018Документ4 страницыList of Certified Seedling Nurseries 2018Kaaya GodfreyОценок пока нет
- A Guide To Kjeldahl Nitrogen Determination Methods and ApparatusДокумент13 страницA Guide To Kjeldahl Nitrogen Determination Methods and ApparatusNoranisza MahmudОценок пока нет
- Quality Manual Body 03Документ32 страницыQuality Manual Body 03Kaaya GodfreyОценок пока нет
- How To Construct A Smoker For BeekeepingДокумент8 страницHow To Construct A Smoker For BeekeepingRicardoОценок пока нет
- Electrode Kinetics: MCH 7105: Advances in ElectrochmistryДокумент25 страницElectrode Kinetics: MCH 7105: Advances in ElectrochmistryKaaya GodfreyОценок пока нет
- V CollectДокумент1 страницаV CollectKaaya GodfreyОценок пока нет
- Bore Hole 1 (Sr. No.23259) Daily Water ConsumptionДокумент1 страницаBore Hole 1 (Sr. No.23259) Daily Water ConsumptionKaaya GodfreyОценок пока нет
- Kyambogo UniversityДокумент14 страницKyambogo UniversityKaaya GodfreyОценок пока нет
- MasloДокумент15 страницMasloKaaya GodfreyОценок пока нет
- Quality Chemical HSE PolicyДокумент1 страницаQuality Chemical HSE PolicyKaaya GodfreyОценок пока нет
- Jo PolymersДокумент17 страницJo PolymersKaaya GodfreyОценок пока нет
- Environmental WorkДокумент81 страницаEnvironmental WorkKaaya GodfreyОценок пока нет
- Equal Remuneration ActДокумент17 страницEqual Remuneration ActRohit SawaleОценок пока нет
- Jacky VEDCO PDFДокумент1 страницаJacky VEDCO PDFKaaya GodfreyОценок пока нет
- Kyambogo FeesДокумент1 страницаKyambogo FeesKaaya GodfreyОценок пока нет
- Ohse Training Invitation at UmaДокумент4 страницыOhse Training Invitation at UmaKaaya GodfreyОценок пока нет
- Science 315Документ22 страницыScience 315Kaaya GodfreyОценок пока нет
- Formative Research Report - FinalДокумент31 страницаFormative Research Report - FinalKaaya GodfreyОценок пока нет
- Science 315Документ22 страницыScience 315Kaaya GodfreyОценок пока нет
- To BeДокумент1 страницаTo BeKaaya GodfreyОценок пока нет
- Task: Assignment 1 Lecturer: Mr. Mogany Moses Year: Yr Iii Semister: IДокумент8 страницTask: Assignment 1 Lecturer: Mr. Mogany Moses Year: Yr Iii Semister: IKaaya GodfreyОценок пока нет
- Industrial Training Report Submitted in Partial Fulfillment of The Requirements For The Award of The D ProcurementДокумент2 страницыIndustrial Training Report Submitted in Partial Fulfillment of The Requirements For The Award of The D ProcurementKaaya GodfreyОценок пока нет
- Uganda Land Policy Final Draft 30 March 20112 PDFДокумент61 страницаUganda Land Policy Final Draft 30 March 20112 PDFKaaya GodfreyОценок пока нет
- Essential Log Books for Plant Security and OperationsДокумент7 страницEssential Log Books for Plant Security and OperationsKaaya GodfreyОценок пока нет
- Industrial RelationsДокумент8 страницIndustrial RelationsKaaya GodfreyОценок пока нет
- Not YetДокумент1 страницаNot YetKaaya GodfreyОценок пока нет
- Essential Log Books for Plant Security and OperationsДокумент7 страницEssential Log Books for Plant Security and OperationsKaaya GodfreyОценок пока нет
- Online Graduate Recruitment FormДокумент4 страницыOnline Graduate Recruitment FormKaaya GodfreyОценок пока нет
- CHMT 3037 TutorialsДокумент2 страницыCHMT 3037 TutorialsAbdulsalam JibrilОценок пока нет
- MCQ For Class 12 Chapterwise PDFДокумент266 страницMCQ For Class 12 Chapterwise PDFAgape Sol'ns85% (13)
- ECUST PROII Advanced Training PDFДокумент118 страницECUST PROII Advanced Training PDFframon.chem35Оценок пока нет
- Phenol ProductionДокумент9 страницPhenol ProductionPlant Design100% (1)
- 9 Chapter Solutions Short Questions With AnswerДокумент8 страниц9 Chapter Solutions Short Questions With AnswerBilal KhanОценок пока нет
- Process Simulation Essentials - Example BookДокумент63 страницыProcess Simulation Essentials - Example BookRebeca LópezОценок пока нет
- Chapter 3Документ61 страницаChapter 3rejie magnayeОценок пока нет
- Determination of Alcohol Content in Alcoholic BeveragesДокумент7 страницDetermination of Alcohol Content in Alcoholic BeveragesKaye Danielle HilomenОценок пока нет
- Fractional Distillation of Ginebra San Miguel GinДокумент6 страницFractional Distillation of Ginebra San Miguel GinHajime NakaegawaОценок пока нет
- Emerson Webinar on Refrigerant Glide EffectsДокумент26 страницEmerson Webinar on Refrigerant Glide EffectspaldopalОценок пока нет
- Veolia Case StudyДокумент15 страницVeolia Case StudyKanakarao MalappareddyОценок пока нет
- EP000937029B1 Process for Ethyl Acetate Production from Ethanol OxidationДокумент9 страницEP000937029B1 Process for Ethyl Acetate Production from Ethanol OxidationMuhammad Yanuar AnantaОценок пока нет
- Chemical Engineering - 1999 Section A (75 Marks) ExamДокумент13 страницChemical Engineering - 1999 Section A (75 Marks) ExamAnonymous 8pCXXsОценок пока нет
- Azo Sep - Company Selling Pervap Technique PDFДокумент31 страницаAzo Sep - Company Selling Pervap Technique PDFAkhil AggarwalОценок пока нет
- Reactive DistillationДокумент8 страницReactive DistillationSalim ChohanОценок пока нет
- Alternative Techniques For Defatting Soy - A Practical ReviewДокумент24 страницыAlternative Techniques For Defatting Soy - A Practical ReviewCristian Mateo Ovalle CifuentesОценок пока нет
- Ester Reactions of Fatty Acids PDFДокумент12 страницEster Reactions of Fatty Acids PDFGrecia SuffoОценок пока нет
- Simulation and Analysis of A Reactive Distillation Column For Removal of Water From Ethanol Water MixturesДокумент9 страницSimulation and Analysis of A Reactive Distillation Column For Removal of Water From Ethanol Water MixturesBryanJianОценок пока нет
- ChemДокумент4 страницыChemishitwa mishraОценок пока нет
- Moonlight & Roses PDFДокумент63 страницыMoonlight & Roses PDFTitu A FaОценок пока нет
- Tutorial 5 Phase EquilibriumДокумент3 страницыTutorial 5 Phase EquilibriumezanaОценок пока нет
- 2009 12 Brouwer UreaKnowHow - Com Phase Diagrams of The Urea ProcessДокумент11 страниц2009 12 Brouwer UreaKnowHow - Com Phase Diagrams of The Urea ProcessjunaidОценок пока нет
- CET II (3140507) Study MaterialДокумент37 страницCET II (3140507) Study MaterialAkasH BinDОценок пока нет
- CH 7 Assignment PDFДокумент3 страницыCH 7 Assignment PDFAftab57.Оценок пока нет
- Distillation: An IntroductionДокумент24 страницыDistillation: An IntroductionDozdi100% (1)
- Enthalpy vs. Composition - Ponchon-Savarit PlotДокумент32 страницыEnthalpy vs. Composition - Ponchon-Savarit PlotahmedОценок пока нет
- Lecture Notes: Subject: Chemistry-III (Basic Physical-I) Faculty: Dr. Monalisa Mohapatra Subject Code: CH-211Документ103 страницыLecture Notes: Subject: Chemistry-III (Basic Physical-I) Faculty: Dr. Monalisa Mohapatra Subject Code: CH-211Let's FunОценок пока нет
- Chem 343 Experiment 3 Lab ReportДокумент7 страницChem 343 Experiment 3 Lab ReportAddison GasserОценок пока нет
- Artículo Ethyl AcetateДокумент13 страницArtículo Ethyl AcetateSebastianOlayaGomezОценок пока нет