Вам также может понравиться
- Quick Revision of Bio Phy Che 9 HoursДокумент489 страницQuick Revision of Bio Phy Che 9 Hourscbsegirlsaipmt100% (2)
- Samuel Glasstone - Thermodynamics For Chemists PDFДокумент532 страницыSamuel Glasstone - Thermodynamics For Chemists PDFRimmon Singh100% (2)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (890)
- Image ProcessingДокумент49 страницImage ProcessingJyoti SinghОценок пока нет
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Egee 101 Reflective Essay 1Документ3 страницыEgee 101 Reflective Essay 1api-142590237Оценок пока нет
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- The Synchronus Rotor Instability Phenomenon - Morton Effect PDFДокумент9 страницThe Synchronus Rotor Instability Phenomenon - Morton Effect PDFabdullah buttОценок пока нет
- Computational Methods For Platicity-SouzaДокумент816 страницComputational Methods For Platicity-SouzaMel Santos100% (7)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- Amateur's Telescope Was First Published in 1920. However, Unlike Ellison's TimeДокумент4 страницыAmateur's Telescope Was First Published in 1920. However, Unlike Ellison's Timemohamadazaresh0% (1)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- A Low Order System Frequency Response ModelДокумент10 страницA Low Order System Frequency Response ModelNadil AminОценок пока нет
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- Comparing Masses of Reactants and ProductsДокумент4 страницыComparing Masses of Reactants and ProductsDaniel TriumbariОценок пока нет
- Spectrophotometric Determination of The Equilibrium Constant of A ReactionДокумент5 страницSpectrophotometric Determination of The Equilibrium Constant of A Reactionnarras11100% (1)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- CHE572 Chapter 2 Particle Size Characterization PDFДокумент18 страницCHE572 Chapter 2 Particle Size Characterization PDFMuhd FahmiОценок пока нет
- Electrical Engineering BA (B), Analog Electronics, ET065G 6 Credits ET064G 7.5 CreditsДокумент43 страницыElectrical Engineering BA (B), Analog Electronics, ET065G 6 Credits ET064G 7.5 Creditsdev-nullОценок пока нет
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- Britishhomoeopat 00 BritialaДокумент448 страницBritishhomoeopat 00 BritialaAlbena Trifonova0% (2)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- Reciprocating compressor performance analysis using computer simulationДокумент9 страницReciprocating compressor performance analysis using computer simulationLeandro Garcia VelaОценок пока нет
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2219)
- 1konsep Dasar TermodinamikaДокумент39 страниц1konsep Dasar TermodinamikamisrakandiОценок пока нет
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- 1 Electricity Test QuestionsДокумент5 страниц1 Electricity Test QuestionstinoОценок пока нет
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- ROCKYДокумент4 страницыROCKYNelson Alexander Aponte SimbronОценок пока нет
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (265)
- The Basic Differential Equation For Radial Flow in A Porous MediumДокумент8 страницThe Basic Differential Equation For Radial Flow in A Porous MediumrestofficalОценок пока нет
- Teknik Menjawab KIMIA 2011Документ73 страницыTeknik Menjawab KIMIA 2011Nur HakimОценок пока нет
- How Does DCPIP WorkДокумент3 страницыHow Does DCPIP WorkIsaac LeeОценок пока нет
- PCAB List of Licensed Contractors For CFY 2017-2018 As of 19 Sep 2017 - Web2Документ175 страницPCAB List of Licensed Contractors For CFY 2017-2018 As of 19 Sep 2017 - Web2Paige Lim76% (21)
- Journal of Power Sources: Pedro O. Lopez-Montesinos, Amit V. Desai, Paul J.A. KenisДокумент8 страницJournal of Power Sources: Pedro O. Lopez-Montesinos, Amit V. Desai, Paul J.A. KenisbernardОценок пока нет
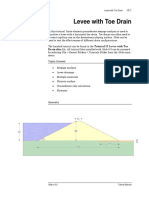
- Levee Drain Analysis in SlideДокумент12 страницLevee Drain Analysis in SlideAdriRGОценок пока нет
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Mystery of Relationship Charts RevealedДокумент5 страницThe Mystery of Relationship Charts RevealedPhalgun Balaaji0% (1)
- Offshore Pipeline Hydraulic and Mechanical AnalysesДокумент25 страницOffshore Pipeline Hydraulic and Mechanical AnalysesEslam RedaОценок пока нет
- Partition Coefficients and Their UsesДокумент92 страницыPartition Coefficients and Their UsesquelenigОценок пока нет
- LNG SamplingSystemДокумент2 страницыLNG SamplingSystemGuillermo Lopez-FloresОценок пока нет
- A First Course in Optimization Theory - ContentДокумент8 страницA First Course in Optimization Theory - ContentSuraj KumarОценок пока нет
- CNC CONTOUR VALIDATION WITH LASER BALL BARДокумент15 страницCNC CONTOUR VALIDATION WITH LASER BALL BARNitin DaduОценок пока нет
- Hammer, Harper, Ryan - 2001 - Past Paleontological Statistics Software Package For Education and Data AnalysisДокумент7 страницHammer, Harper, Ryan - 2001 - Past Paleontological Statistics Software Package For Education and Data AnalysisA3A31234Оценок пока нет
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (119)