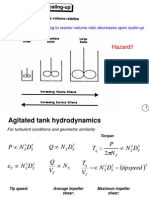
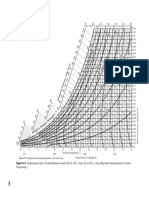
Вам также может понравиться
- Aeration and AgitationДокумент9 страницAeration and AgitationKasun Prasanna SilvaОценок пока нет
- Bioreactor ModelingДокумент28 страницBioreactor ModelingShailendra Singh Khichi100% (1)
- Mechanical Biological & Chemical (MBC) Treatment Plant: Collection of Waste WaterДокумент4 страницыMechanical Biological & Chemical (MBC) Treatment Plant: Collection of Waste Watervinay ChalagundlaОценок пока нет
- Unit Operation Laboratory ProtocolДокумент73 страницыUnit Operation Laboratory ProtocolSreemoyee ChakrabortyОценок пока нет
- Ideal Bioreactor EngineeringДокумент60 страницIdeal Bioreactor EngineeringAyesha RalliyaОценок пока нет
- Phosgene Micro Reactor A I CheДокумент9 страницPhosgene Micro Reactor A I CheJorge RamirezОценок пока нет
- Continuous Fermentation of Sago Starch to Solvent Using C. saccharobutylicumДокумент9 страницContinuous Fermentation of Sago Starch to Solvent Using C. saccharobutylicumputrianneОценок пока нет
- Advanced Process Control and DynamicsДокумент273 страницыAdvanced Process Control and DynamicsYinka OlatunjiОценок пока нет
- Stoichiometry of Microbial Growth and Product FormationДокумент30 страницStoichiometry of Microbial Growth and Product FormationMark GerardОценок пока нет
- Methanol From GlycerinДокумент5 страницMethanol From GlycerinaudreysosoОценок пока нет
- Oxygen Transfer RateДокумент11 страницOxygen Transfer RatefayeОценок пока нет
- Mass Transfer Sample ChaptersДокумент29 страницMass Transfer Sample ChaptersGurunath EpiliОценок пока нет
- Sathyabama University Mass Transfer Lab ManualДокумент60 страницSathyabama University Mass Transfer Lab ManualAhmed AliОценок пока нет
- Plate & Frame Heat ExchangerДокумент20 страницPlate & Frame Heat Exchangerzohaib sattarОценок пока нет
- RI Vs Composition Methanol-Water MixtureДокумент12 страницRI Vs Composition Methanol-Water MixtureAnonymous VeJYFSMWLIОценок пока нет
- Process Description of Paracetamol ManufacturingДокумент25 страницProcess Description of Paracetamol ManufacturingAllan ChongОценок пока нет
- Convergence Hints (Aspen)Документ13 страницConvergence Hints (Aspen)Saurabh GuptaОценок пока нет
- Chemical Engineering - 1999 Section A (75 Marks) ExamДокумент13 страницChemical Engineering - 1999 Section A (75 Marks) ExamAnonymous 8pCXXsОценок пока нет
- ChE-433 Reaction Engineering Lab ManualДокумент25 страницChE-433 Reaction Engineering Lab ManualHasan AkhuamariОценок пока нет
- Solutions Manual Unit Operations of ChemДокумент19 страницSolutions Manual Unit Operations of ChemPutri AdantiОценок пока нет
- Mechanical Unit OperationsДокумент8 страницMechanical Unit OperationsFA AyОценок пока нет
- SCALE UP FERMENTATION PROCESSESДокумент11 страницSCALE UP FERMENTATION PROCESSESLouella100% (1)
- Isothermal ReactorДокумент58 страницIsothermal ReactorRoxanna LevineОценок пока нет
- Effect of Mass TransferДокумент5 страницEffect of Mass Transferengr20210% (1)
- Microbial Growth Kinetics and BioreactorsДокумент10 страницMicrobial Growth Kinetics and BioreactorsKareem TarekОценок пока нет
- Reactor Selection CriteriaДокумент2 страницыReactor Selection CriteriaJunaid JohnsonОценок пока нет
- Cation-Exchange Chromatography - An Overview - ScienceDirect TopicsДокумент12 страницCation-Exchange Chromatography - An Overview - ScienceDirect TopicsBharath BhoseОценок пока нет
- 39 Algal Oil Production 1Документ21 страница39 Algal Oil Production 1Sai Srivathsava UdathuОценок пока нет
- Rr410802 Chemical Reaction Engineering IIДокумент8 страницRr410802 Chemical Reaction Engineering IISrinivasa Rao G100% (3)
- PDFДокумент74 страницыPDFHARISHKOTHARU48Оценок пока нет
- Residence - Time DistributionДокумент9 страницResidence - Time DistributionNik Nur Izzatul IkmalОценок пока нет
- Process Intensification ReportДокумент12 страницProcess Intensification ReportSushma SusmitaОценок пока нет
- CHE4162 Particle Technology: Chemical Reactions in Fluidized BedsДокумент27 страницCHE4162 Particle Technology: Chemical Reactions in Fluidized BedsPhan NeeОценок пока нет
- Biogas PostaДокумент83 страницыBiogas PostaMarcelo TorresОценок пока нет
- C H O + A O + B NH C C H NO + D H O+eCO: InstructionsДокумент4 страницыC H O + A O + B NH C C H NO + D H O+eCO: InstructionsJohn Paul Jandayan33% (3)
- DSTWU - A Shortcut Distillation Model in Aspen Plus V8.0Документ11 страницDSTWU - A Shortcut Distillation Model in Aspen Plus V8.0JúpiterОценок пока нет
- An Industrial Wastewater Contains 10 MG L ChlorophenolДокумент5 страницAn Industrial Wastewater Contains 10 MG L ChlorophenolsumitОценок пока нет
- Richard P Feynman-Surely Youre Joking MR Feynman v5Документ3 страницыRichard P Feynman-Surely Youre Joking MR Feynman v5Anonymous Nayak0% (1)
- Chemical Process Design: Overview of Process Synthesis and Generation of FlowsheetsДокумент19 страницChemical Process Design: Overview of Process Synthesis and Generation of Flowsheetsxhche7Оценок пока нет
- PHE Control Mechanism PDFДокумент6 страницPHE Control Mechanism PDFNaren VmdОценок пока нет
- GTU Mass Transfer OperationsДокумент4 страницыGTU Mass Transfer Operationslata sinsinwarОценок пока нет
- General Guidelines For Distillation ColumnДокумент23 страницыGeneral Guidelines For Distillation ColumnCristinaОценок пока нет
- OTK 1 Lecture1#AgitationMixingДокумент36 страницOTK 1 Lecture1#AgitationMixingKevin EsmunaldoОценок пока нет
- Lecture Notes-Bioreactor Design and Operation-1Документ19 страницLecture Notes-Bioreactor Design and Operation-1lazytinku100% (1)
- CG5052 - Agitation and Mixing - 2 - 2015 PDFДокумент32 страницыCG5052 - Agitation and Mixing - 2 - 2015 PDFzuopengxiangОценок пока нет
- Crystallizers and Their ControlДокумент34 страницыCrystallizers and Their ControlVinitaVartakОценок пока нет
- Erasmus Mass 2004 PDFДокумент257 страницErasmus Mass 2004 PDFSherLockОценок пока нет
- Laboratory Scale Water Circuit Including A Photocatalytic ReactorДокумент8 страницLaboratory Scale Water Circuit Including A Photocatalytic ReactorPatrick_NickelsОценок пока нет
- Catalytic Hydrogenation in The Liquid PhaseДокумент8 страницCatalytic Hydrogenation in The Liquid PhasegiovanniОценок пока нет
- Experiment No 18Документ4 страницыExperiment No 18Suvrasoumya Mohanty100% (2)
- National University Process Dynamics Modelling & Control CourseДокумент15 страницNational University Process Dynamics Modelling & Control CourseKundayi ChagwederaОценок пока нет
- Continuous Process Technology A Tool ForДокумент8 страницContinuous Process Technology A Tool ForAmjadRiazОценок пока нет
- Batch Reactor: Optical High Precision Components by Hellma in The European Columbus Space LaboratoryДокумент16 страницBatch Reactor: Optical High Precision Components by Hellma in The European Columbus Space Laboratoryangelie_gubatanОценок пока нет
- Plug Flow Reactor ExperimentДокумент16 страницPlug Flow Reactor ExperimentN Afiqah RazakОценок пока нет
- 5.2. Classification of FuelsДокумент16 страниц5.2. Classification of FuelsadiОценок пока нет
- Lab ManualДокумент63 страницыLab ManualRianna SОценок пока нет
- Plant Design of Acetone ProductionДокумент20 страницPlant Design of Acetone ProductionMary Grace VelitarioОценок пока нет
- Ionic Liquids in Lipid Processing and Analysis: Opportunities and ChallengesОт EverandIonic Liquids in Lipid Processing and Analysis: Opportunities and ChallengesXuebing XuОценок пока нет
- Insights into Chemical Engineering: Selected Papers of P.V. DanckwertsОт EverandInsights into Chemical Engineering: Selected Papers of P.V. DanckwertsОценок пока нет
- Apex UltimaДокумент2 страницыApex UltimavijaygovindarajОценок пока нет
- OM Research Fellowship JRF SRF RAДокумент2 страницыOM Research Fellowship JRF SRF RAvijaygovindarajОценок пока нет
- (Histag Not Removed) Potent Delivery of Functional Proteins Into Mammalian Cells in Vitro and in Vivo Using A Supercharged ProteinДокумент6 страниц(Histag Not Removed) Potent Delivery of Functional Proteins Into Mammalian Cells in Vitro and in Vivo Using A Supercharged ProteinvijaygovindarajОценок пока нет
- Terms and ConditionsДокумент5 страницTerms and ConditionsvijaygovindarajОценок пока нет
- Form 30Документ2 страницыForm 30Sriraghuraman Gopal Rathnam0% (1)
- Medical Certificate of FitnessДокумент1 страницаMedical Certificate of FitnessvijaygovindarajОценок пока нет
- Coomassie StainingДокумент1 страницаCoomassie StainingvijaygovindarajОценок пока нет
- Dissertation TemplateДокумент17 страницDissertation TemplateDeep JoshiОценок пока нет
- Tata Innovation Fellowship 2012Документ2 страницыTata Innovation Fellowship 2012vijaygovindarajОценок пока нет
- RSC Preparation of Ammonium Sulfate Student PDFДокумент1 страницаRSC Preparation of Ammonium Sulfate Student PDFextraОценок пока нет
- Summary Notes Waste LandДокумент14 страницSummary Notes Waste LandRozi KhanОценок пока нет
- CalendarДокумент1 страницаCalendarvijaygovindarajОценок пока нет
- SEM Sample Preparation Cultured CellsДокумент2 страницыSEM Sample Preparation Cultured CellsvijaygovindarajОценок пока нет
- 4% PFA (Paraformaldehyde) - ML: 4% Paraformaldehyde in 1 X Phosphate Buffered Saline - PH 7.4Документ1 страница4% PFA (Paraformaldehyde) - ML: 4% Paraformaldehyde in 1 X Phosphate Buffered Saline - PH 7.4vijaygovindarajОценок пока нет
- Christian Medical College, VelloreДокумент1 страницаChristian Medical College, VellorevijaygovindarajОценок пока нет
- Methyl at Ion Specific PCR A Novel PCR Assay For Methyl at IonДокумент6 страницMethyl at Ion Specific PCR A Novel PCR Assay For Methyl at IonRenata BarbosaОценок пока нет
- IPGphor II ManualДокумент35 страницIPGphor II ManualvijaygovindarajОценок пока нет
- CCD Camera LEI-5.0-CD Sensor SpecsДокумент1 страницаCCD Camera LEI-5.0-CD Sensor SpecsvijaygovindarajОценок пока нет
- PRSETc SLPIДокумент2 страницыPRSETc SLPIvijaygovindarajОценок пока нет
- 5th Proteomics Scientific ScheduleДокумент3 страницы5th Proteomics Scientific SchedulevijaygovindarajОценок пока нет
- 5th Proteomics Scientific ScheduleДокумент3 страницы5th Proteomics Scientific SchedulevijaygovindarajОценок пока нет
- Protein Estimation by Lawry's Method: Unknown SamplesДокумент3 страницыProtein Estimation by Lawry's Method: Unknown SamplesvijaygovindarajОценок пока нет
- TR0049 Acetone PrecipitationДокумент2 страницыTR0049 Acetone PrecipitationvijaygovindarajОценок пока нет
- List of Govt Holidays of 2012Документ6 страницList of Govt Holidays of 2012makarandphage@yahoo.co.inОценок пока нет
- Quantification of Healthy Follicles in The Neonatal and Adult Mouse OvaryДокумент15 страницQuantification of Healthy Follicles in The Neonatal and Adult Mouse OvaryvijaygovindarajОценок пока нет
- Lo Wry ProcedureДокумент1 страницаLo Wry ProcedurevijaygovindarajОценок пока нет
- อัตราภาษีของไทยที่ลดให้เปรูДокумент124 страницыอัตราภาษีของไทยที่ลดให้เปรูDante FilhoОценок пока нет
- Quant One Analyser – endless possibilitiesДокумент6 страницQuant One Analyser – endless possibilitiesSamuel SuОценок пока нет
- Features General Description: 3A 24V 340Khz Synchronous Buck ConverterДокумент18 страницFeatures General Description: 3A 24V 340Khz Synchronous Buck ConverterAntonioNobregaОценок пока нет
- Overview of Pathophysiology of Hypoxemia and HypoxiaДокумент15 страницOverview of Pathophysiology of Hypoxemia and HypoxiaMARY ANN CAGATANОценок пока нет
- MACRO-ETCHING SOLUTIONS FOR ALUMINIUM ALLOYSДокумент1 страницаMACRO-ETCHING SOLUTIONS FOR ALUMINIUM ALLOYSsensoham03Оценок пока нет
- Mar For M: I MMQ SeriesДокумент28 страницMar For M: I MMQ SeriesIpal Febri NartaОценок пока нет
- Parts of Speech 15Документ16 страницParts of Speech 15lost finОценок пока нет
- 322439480MVR Single Page Single Page Booklet - OPTДокумент12 страниц322439480MVR Single Page Single Page Booklet - OPTlarry vargas bautistaОценок пока нет
- Dokumen - Tips - Astm A535 9 Percent NickelДокумент5 страницDokumen - Tips - Astm A535 9 Percent NickelJeovanne CabralОценок пока нет
- Dimensions and Relations of The Dentogingival Junction in Humans. Gargiulo 1961Документ7 страницDimensions and Relations of The Dentogingival Junction in Humans. Gargiulo 1961Linda Garcia PОценок пока нет
- English Test 6Документ87 страницEnglish Test 6Ha PhanОценок пока нет
- Turkey ImportДокумент14 страницTurkey ImportMani 1Оценок пока нет
- Plant LayoutДокумент16 страницPlant LayoutAli MahmoudОценок пока нет
- Fe in Black TeaДокумент6 страницFe in Black TeaHerni Nur AeniОценок пока нет
- Technical PaperДокумент6 страницTechnical PaperSIJO MONACHANОценок пока нет
- Gold Grade of Epithermal Gold Ore at Lamuntet, Brang Rea, West Sumbawa District, West Nusa Tenggara Province, IndonesiaДокумент10 страницGold Grade of Epithermal Gold Ore at Lamuntet, Brang Rea, West Sumbawa District, West Nusa Tenggara Province, Indonesiasukri arjunaОценок пока нет
- HistorydylaneditДокумент6 страницHistorydylaneditapi-19858424Оценок пока нет
- VGHV NBV GH fc7fvbn BN NGCJHGДокумент16 страницVGHV NBV GH fc7fvbn BN NGCJHGRahul GОценок пока нет
- Pd3c CV Swa (22kv)Документ2 страницыPd3c CV Swa (22kv)เต่า วีไอОценок пока нет
- ASSEMBLING COMPUTER: HOW TO BUILD A PCДокумент48 страницASSEMBLING COMPUTER: HOW TO BUILD A PCCeejaay PelinaОценок пока нет
- Carta Psicrometrica PDFДокумент2 страницыCarta Psicrometrica PDFJuliethОценок пока нет
- M. Valerio Assignment 6.1Документ1 страницаM. Valerio Assignment 6.1Mark Kristian ValerioОценок пока нет
- 1B Cosmos-Standard - Technical - Guide - v40Документ45 страниц1B Cosmos-Standard - Technical - Guide - v40carla deiddaОценок пока нет
- Horizontal Projectile WSДокумент3 страницыHorizontal Projectile WSForsbergPhysicsОценок пока нет
- Geotechnical Elements and Models in OpenSeesДокумент21 страницаGeotechnical Elements and Models in OpenSeesUmut AkınОценок пока нет
- 2nd - Science-Second-Quarter-Week-1Документ37 страниц2nd - Science-Second-Quarter-Week-1Arlene AranzasoОценок пока нет
- Synchronized Natural Incubation by Free-Range Native ChickensДокумент2 страницыSynchronized Natural Incubation by Free-Range Native ChickensFilbert John MillanОценок пока нет
- Hobby 01: COD. 9942062.01 REV. 00Документ9 страницHobby 01: COD. 9942062.01 REV. 00Alexander SharamiginОценок пока нет
- Paradise Pools Flyer With Price ListДокумент5 страницParadise Pools Flyer With Price ListKhuzaima HussainОценок пока нет
- Common Sense Mechanics 9Документ9 страницCommon Sense Mechanics 9Vikas VatsОценок пока нет