Вам также может понравиться
- Police Guide To Illegally Concealed WeaponsДокумент171 страницаPolice Guide To Illegally Concealed Weaponsdirectvguy6969100% (4)
- Building A Button Rifling Machine by Harold HoffmanДокумент15 страницBuilding A Button Rifling Machine by Harold HoffmanNorm100% (18)
- LIBARATOR Prints PDFДокумент31 страницаLIBARATOR Prints PDFstilmix60Оценок пока нет
- 05 13 04 ATF LTR Making WeaponsДокумент2 страницы05 13 04 ATF LTR Making Weaponsstilmix60Оценок пока нет
- 05 13 04 ATF LTR Making WeaponsДокумент2 страницы05 13 04 ATF LTR Making Weaponsstilmix60Оценок пока нет
- John G. Zimmerman Master Gunsmith PDFДокумент43 страницыJohn G. Zimmerman Master Gunsmith PDFstilmix60Оценок пока нет
- Mil STD 1913 SpecsДокумент11 страницMil STD 1913 SpecsMarcus DragoОценок пока нет
- 04 02 04 ATF LTR LEO Stamped LowersДокумент1 страница04 02 04 ATF LTR LEO Stamped Lowersstilmix60Оценок пока нет
- 8 29 05 Receiver WeldsДокумент2 страницы8 29 05 Receiver Weldsstilmix60Оценок пока нет
- 05 25 04 ATF LTR Manufacture of WeaponsДокумент2 страницы05 25 04 ATF LTR Manufacture of Weaponsstilmix60Оценок пока нет
- 04 02 04 ATF LTR LEO Stamped LowersДокумент1 страница04 02 04 ATF LTR LEO Stamped Lowersstilmix60Оценок пока нет
- 05 25 04 ATF LTR Manufacture of WeaponsДокумент2 страницы05 25 04 ATF LTR Manufacture of Weaponsstilmix60Оценок пока нет
- 2563 The Model EngineerДокумент39 страниц2563 The Model Engineerstilmix60100% (1)
- Installing the adjuster plate and jackshaft assemblyДокумент31 страницаInstalling the adjuster plate and jackshaft assemblystilmix60100% (2)
- 2 10 05 M16 Carrier AR15Документ2 страницы2 10 05 M16 Carrier AR15stilmix60Оценок пока нет
- 2556 The Model EngineerДокумент36 страниц2556 The Model Engineerstilmix60100% (1)
- Homemade Black PowderДокумент5 страницHomemade Black Powderstilmix60100% (1)
- SulfurДокумент2 страницыSulfurstilmix60100% (1)
- 2558 The Model EngineerДокумент38 страниц2558 The Model Engineerstilmix60100% (2)
- Homemade Black PowderДокумент5 страницHomemade Black Powderstilmix60100% (1)
- 2562 The Model EngineerДокумент31 страница2562 The Model Engineerstilmix60Оценок пока нет
- SaltpeterДокумент4 страницыSaltpeterstilmix60100% (1)
- 2561 The Model EngineerДокумент41 страница2561 The Model Engineerstilmix60Оценок пока нет
- 2557 The Model EngineerДокумент39 страниц2557 The Model Engineerstilmix60Оценок пока нет
- 2560 The Model EngineerДокумент40 страниц2560 The Model Engineerstilmix60Оценок пока нет
- Pub 81 Designing Aluminium Bronze CastingsДокумент22 страницыPub 81 Designing Aluminium Bronze Castingsstilmix60Оценок пока нет
- 2559 The Model EngineerДокумент38 страниц2559 The Model Engineerstilmix60100% (1)
- Miyamoto Musashi - A Book of Five Rings Cd2 Id1598746222 Size70Документ35 страницMiyamoto Musashi - A Book of Five Rings Cd2 Id1598746222 Size70fsolomonОценок пока нет
- Rod on Sailing - Lessons from the SeaДокумент105 страницRod on Sailing - Lessons from the Seadavr1chОценок пока нет
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5783)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (890)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (119)
- Boundary LayerДокумент16 страницBoundary LayerAmaterasu Susanoo TsukuyomiОценок пока нет
- Chapter 10 DC CircuitsДокумент8 страницChapter 10 DC CircuitstheturfkitchenОценок пока нет
- Transport Properties of FerromagnetsДокумент58 страницTransport Properties of FerromagnetsJucesBarroОценок пока нет
- Abrasiveness of Dental CeramicsДокумент15 страницAbrasiveness of Dental CeramicsHarish KhundrakpamОценок пока нет
- IDONIAL. Fracture Mechanics FEM PD5500Документ9 страницIDONIAL. Fracture Mechanics FEM PD5500JulioОценок пока нет
- PT-9 Quick InstallationДокумент1 страницаPT-9 Quick InstallationAdi PrasetyoОценок пока нет
- Physics Investigatory ProjectДокумент15 страницPhysics Investigatory ProjectAnand Pratap SinghОценок пока нет
- Steel Design Problem SetxzДокумент35 страницSteel Design Problem SetxzMiah N. PascualОценок пока нет
- Fatigue Behavior of SMA and HMA MixturesДокумент8 страницFatigue Behavior of SMA and HMA MixturesImraanОценок пока нет
- Collector Efficiency Improvement of Recyclic Double-Pass Sheet-And-Tube Solar Water Heaters With Internal Fins AttachedДокумент10 страницCollector Efficiency Improvement of Recyclic Double-Pass Sheet-And-Tube Solar Water Heaters With Internal Fins AttachedResearcherzОценок пока нет
- MS26 Capitulo 3Документ14 страницMS26 Capitulo 3FranklinОценок пока нет
- AUCHLITE PRINCIPAL ION-EXCHANGERSДокумент2 страницыAUCHLITE PRINCIPAL ION-EXCHANGERSSoumitra BanerjeeОценок пока нет
- Epsi 01 01Документ19 страницEpsi 01 01Hamed TorkiОценок пока нет
- Misapplications of BernoulliДокумент1 страницаMisapplications of BernoullipreetОценок пока нет
- RISER STRESS ANALYSIS SLEEVE REPAIR PID 152Документ2 страницыRISER STRESS ANALYSIS SLEEVE REPAIR PID 152Mahamad Azi Bin IshakОценок пока нет
- Polymer: by Adithiyan. D M.Pharm PharmaceuticsДокумент7 страницPolymer: by Adithiyan. D M.Pharm PharmaceuticsAadhi AndersonОценок пока нет
- 76 77Документ2 страницы76 77lemi celemenОценок пока нет
- TGN-D-02 Fatigue Improvement of Welded StructuresДокумент10 страницTGN-D-02 Fatigue Improvement of Welded StructuresakuntayОценок пока нет
- 9320 Manual - PDF de AmperimetroДокумент37 страниц9320 Manual - PDF de AmperimetroluisОценок пока нет
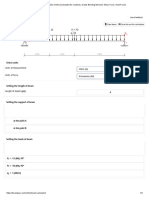
- Beam Calculator Online (Calculate The Reactions, Draws Bending Moment, Shear Force, Axial Force)Документ4 страницыBeam Calculator Online (Calculate The Reactions, Draws Bending Moment, Shear Force, Axial Force)alikhanОценок пока нет
- XRDДокумент26 страницXRDAnonymous dvuYynfXОценок пока нет
- Full Text 01Документ33 страницыFull Text 01Zoric Bobby100% (1)
- Sensors: Metal-Organic Frameworks As Active Materials in Electronic Sensor DevicesДокумент11 страницSensors: Metal-Organic Frameworks As Active Materials in Electronic Sensor Devicespalaniappan_pandianОценок пока нет
- 2022 - JunYe - Sciadv - Hamiltonian Engineering of Spin-Orbit-Coupled Fermions in A Wannier-Stark Optical Lattice ClockДокумент8 страниц2022 - JunYe - Sciadv - Hamiltonian Engineering of Spin-Orbit-Coupled Fermions in A Wannier-Stark Optical Lattice Clock曾许曌秋Оценок пока нет
- Electron ConfigurationДокумент35 страницElectron ConfigurationrayОценок пока нет
- Retrofitting of Reinforced Concrete Beams Using CFRP JacketingДокумент4 страницыRetrofitting of Reinforced Concrete Beams Using CFRP JacketingTaanzОценок пока нет
- Physics Investigatory Project 13 PDF FreeДокумент12 страницPhysics Investigatory Project 13 PDF FreearyanshucalОценок пока нет
- Design of Beam-ColumnsДокумент37 страницDesign of Beam-Columnssalahaddinsharif100% (1)
- This Page Will Be Attached To Your Exams: Physics 7E Formula SheetДокумент1 страницаThis Page Will Be Attached To Your Exams: Physics 7E Formula SheetAhmed ZobiОценок пока нет
- Brass Plate, Sheet, Strip, and Rolled Bar: Standard Specification ForДокумент6 страницBrass Plate, Sheet, Strip, and Rolled Bar: Standard Specification ForOscar OriasОценок пока нет