Вам также может понравиться
- Symmetry Operations 42Документ43 страницыSymmetry Operations 42Parijat BanerjeeОценок пока нет
- The Crystal Structure: A U - STДокумент23 страницыThe Crystal Structure: A U - STMansyur SQeОценок пока нет
- Unit 5Документ19 страницUnit 5Praveen I HiragannavarОценок пока нет
- Crystalline StructureДокумент8 страницCrystalline StructureAdeolu AdelekeОценок пока нет
- Diffuse Field Transmission Into Infinite Sandwich Composite and Laminate Composite CylindersДокумент34 страницыDiffuse Field Transmission Into Infinite Sandwich Composite and Laminate Composite Cylindersmuhammedbusra6716Оценок пока нет
- Research Papers Equivalence of Superspace Groups: Acta Crystallographica Section AДокумент16 страницResearch Papers Equivalence of Superspace Groups: Acta Crystallographica Section Aammayi9845_930467904Оценок пока нет
- Brandon Carter - Brane Dynamics For Treatment of Cosmic Strings and VortonsДокумент89 страницBrandon Carter - Brane Dynamics For Treatment of Cosmic Strings and VortonsJomav23Оценок пока нет
- Review of CrystallographyДокумент20 страницReview of CrystallographyGilda AvendañoОценок пока нет
- Physics of Three-Dimensional Bosonic Topological Insulators: Surface-Deconfined Criticality and Quantized Magnetoelectric EffectДокумент28 страницPhysics of Three-Dimensional Bosonic Topological Insulators: Surface-Deconfined Criticality and Quantized Magnetoelectric Effectcampal123Оценок пока нет
- Part 2. Three Primary Areas of Theoretical ChemistryДокумент73 страницыPart 2. Three Primary Areas of Theoretical ChemistryRyan GoldenОценок пока нет
- Symmetry ElementsДокумент27 страницSymmetry Elementssekar20120Оценок пока нет
- Artigo Porto Normal Mode Determination in CrystalsДокумент38 страницArtigo Porto Normal Mode Determination in CrystalsGelson RodriguesОценок пока нет
- Three-Dimensional Landau Theory Describing The Martensitic Phase Transformation of Shape-Memory AlloysДокумент18 страницThree-Dimensional Landau Theory Describing The Martensitic Phase Transformation of Shape-Memory AlloysMudavath Babu RamОценок пока нет
- Cong JMolBiol 2008Документ6 страницCong JMolBiol 2008huouinkyoumaОценок пока нет
- Alloy: Mixture Metallic Solid Solution Elements Phasemicrostructure HomogeneousДокумент7 страницAlloy: Mixture Metallic Solid Solution Elements Phasemicrostructure HomogeneoussakuraleeshaoranОценок пока нет
- Anisotropy1,2 MBДокумент13 страницAnisotropy1,2 MBRuben AñezОценок пока нет
- 1 2 19examsolДокумент9 страниц1 2 19examsolΦαίδρα ΑμαργιανούОценок пока нет
- Chapter 6. QuasicristalesДокумент48 страницChapter 6. QuasicristalesafnaftanelОценок пока нет
- Organica 4Документ19 страницOrganica 4MarcelaОценок пока нет
- Prediction The Elastic Moduli of CompositesДокумент7 страницPrediction The Elastic Moduli of CompositestraidiaОценок пока нет
- The Influence of Conformational Isomerism On Drug ActionДокумент6 страницThe Influence of Conformational Isomerism On Drug Actionhectorlope45Оценок пока нет
- Cold Atoms in Double-Well Optical Lattices V.I. Yukalov and E.P. YukalovaДокумент24 страницыCold Atoms in Double-Well Optical Lattices V.I. Yukalov and E.P. YukalovaArturo CamachoОценок пока нет
- Basic Concepts of Crystalline StructureДокумент60 страницBasic Concepts of Crystalline StructureKhen Mehko Ojeda100% (1)
- Yidun Wan - Effective Theory of Braid Excitations of Quantum Geometry in Terms of Feynman DiagramsДокумент23 страницыYidun Wan - Effective Theory of Braid Excitations of Quantum Geometry in Terms of Feynman DiagramsMopadDeluxeОценок пока нет
- A Differential Scheme For The Effective Moduli of CompositesДокумент16 страницA Differential Scheme For The Effective Moduli of CompositesAxel DorianОценок пока нет
- Cluster Mergers, Core Oscillations, and Cold FrontsДокумент11 страницCluster Mergers, Core Oscillations, and Cold FrontsEdwin RetanaОценок пока нет
- Inflation Rules, Band Structure, and Localization of Electronic StatesДокумент8 страницInflation Rules, Band Structure, and Localization of Electronic StatesEduardo SaavedraОценок пока нет
- Paper 2Документ9 страницPaper 2Claudio Cofré MansillaОценок пока нет
- Unit - Ii Crystalstructure Analysis: - Analysis of Diffraction Patterns - Inter Planer SpacingДокумент29 страницUnit - Ii Crystalstructure Analysis: - Analysis of Diffraction Patterns - Inter Planer SpacingpariОценок пока нет
- tmp7F1A TMPДокумент9 страницtmp7F1A TMPFrontiersОценок пока нет
- Ssrjuly 2006Документ11 страницSsrjuly 2006andresОценок пока нет
- Symmetry Breaking in Smectics and Surface Models of Their SingularitiesДокумент6 страницSymmetry Breaking in Smectics and Surface Models of Their SingularitiesAnshuman PalОценок пока нет
- PHY323 Statistical MechanicsДокумент66 страницPHY323 Statistical MechanicsKALYESUBULA RONALDОценок пока нет
- Power LawsCSFДокумент16 страницPower LawsCSFc_mc2Оценок пока нет
- Crystal Structure - Wikipedia, The Free EncyclopediaДокумент13 страницCrystal Structure - Wikipedia, The Free EncyclopediaPradeep ChaudhariОценок пока нет
- Class1 - Symmetry Intro - 23102019 - MSRДокумент35 страницClass1 - Symmetry Intro - 23102019 - MSRMANHAR SINGH RAWATОценок пока нет
- Knight 2019Документ11 страницKnight 2019ERIKO DARMAWANОценок пока нет
- MSC Solid State Physics Lecture#4Документ29 страницMSC Solid State Physics Lecture#4Shehzad AhmedОценок пока нет
- Atlas of Atomic Nuclear Structures: Stoyan SargДокумент30 страницAtlas of Atomic Nuclear Structures: Stoyan SargMoniurОценок пока нет
- Numerical Solution Scheme For Inert Disperse and Dilute Gas-Particle FlowsДокумент18 страницNumerical Solution Scheme For Inert Disperse and Dilute Gas-Particle FlowsSungki JungОценок пока нет
- Part of Chapter 2 - Biophysics An IntroductionДокумент6 страницPart of Chapter 2 - Biophysics An IntroductionJmltr RhmhОценок пока нет
- SSP Unit 1Документ44 страницыSSP Unit 1kavithavenkatesanОценок пока нет
- UNIT-4 Stereo-3Документ14 страницUNIT-4 Stereo-3KARTIKAY LADDHAОценок пока нет
- Stereo ChemistryДокумент45 страницStereo Chemistrysanjanaganguly2005Оценок пока нет
- OopsДокумент41 страницаOopsMY MEMORIESОценок пока нет
- Introduction To Q-Tensor TheoryДокумент20 страницIntroduction To Q-Tensor TheoryfivОценок пока нет
- CrystallographyДокумент11 страницCrystallographyShailendra AgarwalОценок пока нет
- M. Meister, F.G. Mertens and A. Sanchez - Soliton Diff Usion On The Classical, Isotropic Heisenberg ChainДокумент13 страницM. Meister, F.G. Mertens and A. Sanchez - Soliton Diff Usion On The Classical, Isotropic Heisenberg Chain23213mОценок пока нет
- Assignment Topics With Materials 1. What Are The Advanced Materials?Документ39 страницAssignment Topics With Materials 1. What Are The Advanced Materials?joshua nithishОценок пока нет
- Ref TBP SP Geom JacsДокумент9 страницRef TBP SP Geom JacsRahul SharmaОценок пока нет
- Fully FinalДокумент48 страницFully Finalalamarif3546Оценок пока нет
- 1 s2.0 0022508878900401 MainДокумент4 страницы1 s2.0 0022508878900401 MainOpenthe DorОценок пока нет
- Systematics of The Spinel Structure TypeДокумент23 страницыSystematics of The Spinel Structure TypeAlin DrucОценок пока нет
- The Structure of Planar Defects in Tilted Perovskites: RichardbeanlandДокумент21 страницаThe Structure of Planar Defects in Tilted Perovskites: RichardbeanlandTran Quang Minh NhatОценок пока нет
- IsomerismДокумент22 страницыIsomerismShivam GuptaОценок пока нет
- Crystal Structure PDFДокумент13 страницCrystal Structure PDFCarolina PardoОценок пока нет
- Nearly Free ElectronДокумент15 страницNearly Free ElectronJohnОценок пока нет
- Rangkuman Materi Crystalline Structure (IBE) Ahmad Naufal Waliyyuddin - 1513623068Документ12 страницRangkuman Materi Crystalline Structure (IBE) Ahmad Naufal Waliyyuddin - 1513623068Ahmad NaufalОценок пока нет
- E4 StereoisomersДокумент6 страницE4 StereoisomersShaun Martel BantuganОценок пока нет
- A Guide To Stress Corrosion Cracking Management EbookДокумент36 страницA Guide To Stress Corrosion Cracking Management Ebookgilbertjerry100% (1)
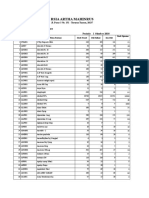
- Rsia Artha Mahinrus: Jl. Pasar 3 No. 151 - Terusan Tuasan, 20237Документ15 страницRsia Artha Mahinrus: Jl. Pasar 3 No. 151 - Terusan Tuasan, 20237Rabyatul Maulida NasutionОценок пока нет
- Artigo Kaolinite Review - Intercalation and Production of PolymerДокумент17 страницArtigo Kaolinite Review - Intercalation and Production of PolymerPhelippe SousaОценок пока нет
- 3.1 WeatheringДокумент25 страниц3.1 WeatheringSipu GiriОценок пока нет
- FastenersДокумент178 страницFastenersthulasi_krishna100% (6)
- ArmaFlex Application UkДокумент36 страницArmaFlex Application UkDave StaelensОценок пока нет
- NSS Chemistry Part 2 The Microscopic World HKCEE Past Paper Question The Microscopic World I Ns - Multiple Choice QuestionsДокумент32 страницыNSS Chemistry Part 2 The Microscopic World HKCEE Past Paper Question The Microscopic World I Ns - Multiple Choice QuestionsミーチェルОценок пока нет
- Tegreen PIP PH 11-2019Документ2 страницыTegreen PIP PH 11-2019Jane Tai100% (1)
- M 10 Dense Graded Stone Mastic Asphalt Specs v5Документ30 страницM 10 Dense Graded Stone Mastic Asphalt Specs v5suhasОценок пока нет
- P70 80 Sealed Filter DriersДокумент11 страницP70 80 Sealed Filter Driersghostz0rОценок пока нет
- Pourbaix DiagramДокумент5 страницPourbaix DiagramChayon MondalОценок пока нет
- Chemical Bonding 2Документ16 страницChemical Bonding 2yvg95100% (1)
- 全棉⾯料(100%Cotton Fabric): Item Comp Yarn Weight (G/SM) Width RemarkДокумент11 страниц全棉⾯料(100%Cotton Fabric): Item Comp Yarn Weight (G/SM) Width RemarkI'm BuddyОценок пока нет
- Casing and Cementing HardwareДокумент4 страницыCasing and Cementing Hardwarezapspaz100% (1)
- Influence of Test Equipment and Procedures On Obtained Accuracy in CPTUДокумент26 страницInfluence of Test Equipment and Procedures On Obtained Accuracy in CPTUalistuguiОценок пока нет
- Water Content and Potential of VegetablesДокумент8 страницWater Content and Potential of VegetablesIsabelle OdenbachОценок пока нет
- Fortification of Biscuits With Fish Protein Concentrate: February 2014Документ9 страницFortification of Biscuits With Fish Protein Concentrate: February 2014syafaiyah dj halidaОценок пока нет
- Geographical Organisation: Marketing ManagerДокумент10 страницGeographical Organisation: Marketing ManagerVinod MalkarОценок пока нет
- Enthusiast Score-I 2021-22Документ1 страницаEnthusiast Score-I 2021-22Aarya Vardhan ShandilyaОценок пока нет
- AccQ Tag SolutionДокумент1 страницаAccQ Tag SolutionNgọc Việt NguyễnОценок пока нет
- Hidroambiente Power Plants 2015dicДокумент6 страницHidroambiente Power Plants 2015dicKoushik DeyОценок пока нет
- Phyto-Mediated Synthesis of Zinc Oxide Nanoparticles of BerberisДокумент31 страницаPhyto-Mediated Synthesis of Zinc Oxide Nanoparticles of BerberisRabeea NasirОценок пока нет
- Material Safety Data Sheets: Product Name: Handy Andy AmmoniaДокумент2 страницыMaterial Safety Data Sheets: Product Name: Handy Andy Ammoniacabeaurey100% (1)
- Lecture 9 - Natural Gas Processing NotesДокумент48 страницLecture 9 - Natural Gas Processing NotesThomas FermatОценок пока нет
- 0570 ChemistryДокумент38 страниц0570 ChemistryLornah LucyОценок пока нет
- In Uence of Bottom Ash and Limestone Powder On The Properties of Ternary Cement and MortarДокумент13 страницIn Uence of Bottom Ash and Limestone Powder On The Properties of Ternary Cement and Mortardummy dumdumОценок пока нет
- TR2000 - Get Datasheet PDFДокумент1 страницаTR2000 - Get Datasheet PDFwendeltrentoОценок пока нет
- Microencapsulation by Spray Drying of Lannea Microcarpa Extract: Technological Characteristics and Antioxidant ActivityДокумент10 страницMicroencapsulation by Spray Drying of Lannea Microcarpa Extract: Technological Characteristics and Antioxidant ActivityJournal of Pharmacy & Pharmacognosy ResearchОценок пока нет
- ABB Technical Note - Cooling Systems - LowresfДокумент2 страницыABB Technical Note - Cooling Systems - LowresfRanjitKumarОценок пока нет
- High Heat Rust Oleum SDSДокумент6 страницHigh Heat Rust Oleum SDSAshish BhanderiОценок пока нет
- Mental Math: How to Develop a Mind for Numbers, Rapid Calculations and Creative Math Tricks (Including Special Speed Math for SAT, GMAT and GRE Students)От EverandMental Math: How to Develop a Mind for Numbers, Rapid Calculations and Creative Math Tricks (Including Special Speed Math for SAT, GMAT and GRE Students)Оценок пока нет
- Quantum Physics: A Beginners Guide to How Quantum Physics Affects Everything around UsОт EverandQuantum Physics: A Beginners Guide to How Quantum Physics Affects Everything around UsРейтинг: 4.5 из 5 звезд4.5/5 (3)
- Build a Mathematical Mind - Even If You Think You Can't Have One: Become a Pattern Detective. Boost Your Critical and Logical Thinking Skills.От EverandBuild a Mathematical Mind - Even If You Think You Can't Have One: Become a Pattern Detective. Boost Your Critical and Logical Thinking Skills.Рейтинг: 5 из 5 звезд5/5 (1)
- Basic Math & Pre-Algebra Workbook For Dummies with Online PracticeОт EverandBasic Math & Pre-Algebra Workbook For Dummies with Online PracticeРейтинг: 4 из 5 звезд4/5 (2)
- Math Workshop, Grade K: A Framework for Guided Math and Independent PracticeОт EverandMath Workshop, Grade K: A Framework for Guided Math and Independent PracticeРейтинг: 5 из 5 звезд5/5 (1)
- Limitless Mind: Learn, Lead, and Live Without BarriersОт EverandLimitless Mind: Learn, Lead, and Live Without BarriersРейтинг: 4 из 5 звезд4/5 (6)
- A Mathematician's Lament: How School Cheats Us Out of Our Most Fascinating and Imaginative Art FormОт EverandA Mathematician's Lament: How School Cheats Us Out of Our Most Fascinating and Imaginative Art FormРейтинг: 5 из 5 звезд5/5 (5)
- Images of Mathematics Viewed Through Number, Algebra, and GeometryОт EverandImages of Mathematics Viewed Through Number, Algebra, and GeometryОценок пока нет
- Mathematical Mindsets: Unleashing Students' Potential through Creative Math, Inspiring Messages and Innovative TeachingОт EverandMathematical Mindsets: Unleashing Students' Potential through Creative Math, Inspiring Messages and Innovative TeachingРейтинг: 4.5 из 5 звезд4.5/5 (21)
- Fluent in 3 Months: How Anyone at Any Age Can Learn to Speak Any Language from Anywhere in the WorldОт EverandFluent in 3 Months: How Anyone at Any Age Can Learn to Speak Any Language from Anywhere in the WorldРейтинг: 3 из 5 звезд3/5 (80)
- ParaPro Assessment Preparation 2023-2024: Study Guide with 300 Practice Questions and Answers for the ETS Praxis Test (Paraprofessional Exam Prep)От EverandParaPro Assessment Preparation 2023-2024: Study Guide with 300 Practice Questions and Answers for the ETS Praxis Test (Paraprofessional Exam Prep)Оценок пока нет