Вам также может понравиться
- International Symposium on Selective Ion-Sensitive Electrodes: International Union of Pure and Applied ChemistryОт EverandInternational Symposium on Selective Ion-Sensitive Electrodes: International Union of Pure and Applied ChemistryG. J. MoodyОценок пока нет
- c1 PDFДокумент43 страницыc1 PDFMadhushan DassanayakeОценок пока нет
- Models For Insulation Aging Under Electrical and Thermal MultistressДокумент12 страницModels For Insulation Aging Under Electrical and Thermal MultistressRogelio RevettiОценок пока нет
- Multiphysics Computational Framework For Cylindrical Lithium-Ion Batteries Under Mechanical Abusive LoadingДокумент13 страницMultiphysics Computational Framework For Cylindrical Lithium-Ion Batteries Under Mechanical Abusive LoadingSravan ChantiОценок пока нет
- Survey of Naval Surface Ship Propulsion and Auxiliary System DevelopmentsДокумент90 страницSurvey of Naval Surface Ship Propulsion and Auxiliary System DevelopmentsAn NgocSonОценок пока нет
- The Load As An Energy Asset in A Distributed DC MGДокумент8 страницThe Load As An Energy Asset in A Distributed DC MGAakanksha DugarОценок пока нет
- Power Control of DC Microgrid With Variable Generation and Energy StorageДокумент5 страницPower Control of DC Microgrid With Variable Generation and Energy StorageSEP-PublisherОценок пока нет
- How To Design Multi-Kw Converters For Electric Vehicles Part 4 - Power Factor Correction PDFДокумент9 страницHow To Design Multi-Kw Converters For Electric Vehicles Part 4 - Power Factor Correction PDFoszemОценок пока нет
- MATLAB-Introduction To ApplicationsДокумент62 страницыMATLAB-Introduction To Applicationsnavz143Оценок пока нет
- Unified Power Quality ConditionerДокумент19 страницUnified Power Quality ConditionerShameer Sr S RОценок пока нет
- Distributed Energy StorageДокумент3 страницыDistributed Energy StoragejuevesОценок пока нет
- Batteries - Fifty Years of Materials DevelopmentДокумент20 страницBatteries - Fifty Years of Materials DevelopmentLucas SerenaОценок пока нет
- Prius Gen IV 1.8L SimulationДокумент14 страницPrius Gen IV 1.8L SimulationseyyidMubeenОценок пока нет
- Power Quality Improvement in Grid Connected PV SystemДокумент6 страницPower Quality Improvement in Grid Connected PV SystemEditor IJTSRDОценок пока нет
- Battery Cooling REPORT V 03 DMДокумент37 страницBattery Cooling REPORT V 03 DMBachir El FilОценок пока нет
- Dynamic Fault Tree AnalysisДокумент24 страницыDynamic Fault Tree AnalysisÖzgürKaymakçıОценок пока нет
- Modelling System Reliability Using Continuous-Time Markov ChainДокумент4 страницыModelling System Reliability Using Continuous-Time Markov ChainjoyopsonОценок пока нет
- What Is A Static SwitchДокумент1 страницаWhat Is A Static SwitchMary HarrisonОценок пока нет
- 19 Induction Motor Fundamentals PDFДокумент37 страниц19 Induction Motor Fundamentals PDFsuchita jainОценок пока нет
- A Cyber-Physical Threat Analysis For MicrogridsДокумент7 страницA Cyber-Physical Threat Analysis For MicrogridsjmescalanteОценок пока нет
- Li-On BatteriesДокумент140 страницLi-On BatteriesShubhОценок пока нет
- Zonal Ship Design (Paper)Документ16 страницZonal Ship Design (Paper)pal_malayОценок пока нет
- Modelling and Simulation of Hybrid Micro Grid Employing DG, PV, Wind and Fuel CellДокумент7 страницModelling and Simulation of Hybrid Micro Grid Employing DG, PV, Wind and Fuel Cellرافع العرفيОценок пока нет
- Voltage and Frequency Control of Inverters Connected in Parallel Forming A Micro-GridДокумент6 страницVoltage and Frequency Control of Inverters Connected in Parallel Forming A Micro-GridShad Rahman100% (1)
- Energy Storage For Power SystemsДокумент124 страницыEnergy Storage For Power SystemsДмитрий Корев0% (1)
- Ac - DC Electronics Laboratory Em-8656Документ126 страницAc - DC Electronics Laboratory Em-8656JonhGonzález100% (1)
- FARS Running Financial Reports and StatisticsДокумент5 страницFARS Running Financial Reports and StatisticsKristen FahyОценок пока нет
- List of Thermal Conductivities - Wikipedia, The Free EncyclopediaДокумент9 страницList of Thermal Conductivities - Wikipedia, The Free EncyclopediaPragasti Nilam SariОценок пока нет
- Batteries & Fuel Cells-1Документ33 страницыBatteries & Fuel Cells-1Abdo MohdyОценок пока нет
- Industrial Load ModelingДокумент15 страницIndustrial Load ModelingSandeepОценок пока нет
- Different Modeling Aspects and Energy Systems of Unified Power Quality Conditioner (UPQC) - An Overview (#168251) - 148354Документ8 страницDifferent Modeling Aspects and Energy Systems of Unified Power Quality Conditioner (UPQC) - An Overview (#168251) - 148354Marshallchiyan PhilipОценок пока нет
- Chemical Review Before Li Ion Batteries PDFДокумент24 страницыChemical Review Before Li Ion Batteries PDFAditya HuseinОценок пока нет
- Supercapacitors 130206061102 Phpapp02Документ22 страницыSupercapacitors 130206061102 Phpapp02Brrijesh YadavОценок пока нет
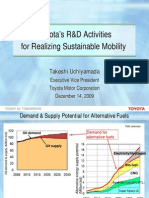
- Toyota's R&D Activities For Realizing Sustainable MobilityДокумент17 страницToyota's R&D Activities For Realizing Sustainable MobilitysamabuelsamidОценок пока нет
- P3T32 en M J006 ANSI Web PDFДокумент426 страницP3T32 en M J006 ANSI Web PDFBruce NationОценок пока нет
- Analysis and Simulation of Photovoltaic Systems Incorporating BatДокумент118 страницAnalysis and Simulation of Photovoltaic Systems Incorporating BatDaniel Gomes JulianoОценок пока нет
- PHD Thesis 20150520 Rev1 Til OrbitДокумент200 страницPHD Thesis 20150520 Rev1 Til OrbitShruti JainОценок пока нет
- Ripple Current Reduction Technique For DC To DC Converter Using Tapped InductorДокумент6 страницRipple Current Reduction Technique For DC To DC Converter Using Tapped InductorInternational Journal of Latest Research in Engineering and TechnologyОценок пока нет
- Modelling and Calculation of The Current Density Distribution Evolution at Vertical Gas-Evolving ElectrodesДокумент17 страницModelling and Calculation of The Current Density Distribution Evolution at Vertical Gas-Evolving ElectrodesmetawfikОценок пока нет
- Lab-On-A-Chip, Microfluidics and Interfacial ElectrokineticsДокумент6 страницLab-On-A-Chip, Microfluidics and Interfacial ElectrokineticsAditya RaghunandanОценок пока нет
- Evaluation of High Power Energy Storage Devices For Use in Compact Pulsed Power SystemsДокумент19 страницEvaluation of High Power Energy Storage Devices For Use in Compact Pulsed Power SystemsbijushresthОценок пока нет
- Professional Integrity and Disobedience in The Military PDFДокумент15 страницProfessional Integrity and Disobedience in The Military PDFLeo BiadnesОценок пока нет
- Potentiostat EIS Setup and OperationДокумент2 страницыPotentiostat EIS Setup and OperationKENT BENEDICT PERALESОценок пока нет
- Large Format Lithium Ion Pouch Cell Full Thermal Characterisation For Improved Electric Vehicle Thermal ManagementДокумент11 страницLarge Format Lithium Ion Pouch Cell Full Thermal Characterisation For Improved Electric Vehicle Thermal ManagementChandrasekaran N100% (1)
- Ultracapacitors (Supercapacitors)Документ21 страницаUltracapacitors (Supercapacitors)NamithaОценок пока нет
- Internal Insulation Failure of HV EquipmentДокумент6 страницInternal Insulation Failure of HV EquipmentashawishОценок пока нет
- The Case For The Diesel EngineДокумент26 страницThe Case For The Diesel EngineChangsun HwangОценок пока нет
- Modelling and Simulation of Power Quality IssuesДокумент27 страницModelling and Simulation of Power Quality IssuesMayaОценок пока нет
- Basics and Growth of Photovoltaic EfficienciesДокумент28 страницBasics and Growth of Photovoltaic EfficienciesMni SdОценок пока нет
- Solar Micro ControllerДокумент18 страницSolar Micro ControllerElangovanОценок пока нет
- PE Power Electronic ConvertersДокумент12 страницPE Power Electronic ConvertersDereje LemmaОценок пока нет
- The Kappa Statistic in Reliability Studies - Use, Interpretation, and Sample Size Requirements - Sim & Wright (2005) PDFДокумент13 страницThe Kappa Statistic in Reliability Studies - Use, Interpretation, and Sample Size Requirements - Sim & Wright (2005) PDFEduardo Aguirre DávilaОценок пока нет
- AS Set Based DesignДокумент150 страницAS Set Based DesignAnonymous yRV1y8KLsОценок пока нет
- Design of Unified Power Quality Conditioner (UPQC)Документ7 страницDesign of Unified Power Quality Conditioner (UPQC)Hari PrasadОценок пока нет
- International Journal of Engineering Research and DevelopmentДокумент8 страницInternational Journal of Engineering Research and DevelopmentIJERDОценок пока нет
- Density of Individual Airborne Wind Energy Systems in AWES FarmsДокумент6 страницDensity of Individual Airborne Wind Energy Systems in AWES FarmsEnrique MartinezОценок пока нет
- Battery ComparisonДокумент7 страницBattery Comparisoninterpon07Оценок пока нет
- Research and Development in Non-Mechanical Electrical Power Sources: Proceedings of the 6th International Symposium Held at Brighton, September 1968От EverandResearch and Development in Non-Mechanical Electrical Power Sources: Proceedings of the 6th International Symposium Held at Brighton, September 1968Оценок пока нет
- New Sensors and Processing ChainОт EverandNew Sensors and Processing ChainJean-Hugh ThomasОценок пока нет
- ME Paper 6 Section 1 PDFДокумент7 страницME Paper 6 Section 1 PDFSaumya SinhaОценок пока нет
- Epicyclic Gear Train Apparatus PDFДокумент1 страницаEpicyclic Gear Train Apparatus PDFSaumya SinhaОценок пока нет
- Automobile Pdf1Документ31 страницаAutomobile Pdf1srinivasnaikОценок пока нет
- Indian Concern Over A US-Pak Nuclear DealДокумент1 страницаIndian Concern Over A US-Pak Nuclear DealSaumya SinhaОценок пока нет
- Me MCQДокумент13 страницMe MCQSaumya SinhaОценок пока нет
- Introduction to Vehicle Transmission ComponentsДокумент21 страницаIntroduction to Vehicle Transmission ComponentsManohara ErlaОценок пока нет
- Advertisement 43 2016 PDFДокумент7 страницAdvertisement 43 2016 PDFSaumya SinhaОценок пока нет
- China Commissions Dam On BrahmaputrДокумент1 страницаChina Commissions Dam On BrahmaputrSaumya SinhaОценок пока нет
- China Ends One Child Policy Time To Dump Malthusian TheoryДокумент2 страницыChina Ends One Child Policy Time To Dump Malthusian TheorySaumya SinhaОценок пока нет
- 91.the Big Picture India-Africa Forum Summit What Is The Agenda PDFДокумент1 страница91.the Big Picture India-Africa Forum Summit What Is The Agenda PDFSaumya SinhaОценок пока нет
- Uniform Civil Code Whats The AgendaДокумент1 страницаUniform Civil Code Whats The AgendaSaumya SinhaОценок пока нет
- China Ends One Child Policy Time To Dump Malthusian TheoryДокумент2 страницыChina Ends One Child Policy Time To Dump Malthusian TheorySaumya SinhaОценок пока нет
- India-Africa Forum Summit What Is The AgendaДокумент1 страницаIndia-Africa Forum Summit What Is The AgendaSaumya SinhaОценок пока нет
- Checking Atrocities Against DalitsДокумент2 страницыChecking Atrocities Against DalitsSaumya SinhaОценок пока нет
- What Caused The Crisis in West AsiaДокумент2 страницыWhat Caused The Crisis in West AsiaSaumya SinhaОценок пока нет
- 24th July MCQДокумент5 страниц24th July MCQSaumya SinhaОценок пока нет
- Ease of Doing Business What Has ChangedДокумент2 страницыEase of Doing Business What Has ChangedSaumya SinhaОценок пока нет
- US-China Face-Off in South China SeaДокумент2 страницыUS-China Face-Off in South China SeaSaumya SinhaОценок пока нет
- 28th July MCQДокумент5 страниц28th July MCQSaumya SinhaОценок пока нет
- India-Nepal Acrimony IncreasesДокумент2 страницыIndia-Nepal Acrimony IncreasesSaumya SinhaОценок пока нет
- Is It Time To Dump Sedition LawДокумент2 страницыIs It Time To Dump Sedition LawSaumya SinhaОценок пока нет
- 30th July MCQДокумент5 страниц30th July MCQSaumya SinhaОценок пока нет
- 26th July 2016 MCQДокумент6 страниц26th July 2016 MCQSaumya SinhaОценок пока нет
- Options For Government On Economic Amp Policy ReformsДокумент1 страницаOptions For Government On Economic Amp Policy ReformsSaumya SinhaОценок пока нет
- Current Affairs Pocket PDF - July 2016 by AffairsCloudДокумент39 страницCurrent Affairs Pocket PDF - July 2016 by AffairsCloudPriyatam BolisettyОценок пока нет
- Current Affairs July 25 2016: Narsingh Yadav Banned from Rio OlympicsДокумент5 страницCurrent Affairs July 25 2016: Narsingh Yadav Banned from Rio OlympicsSaumya SinhaОценок пока нет
- 27th July 2016 MCQДокумент5 страниц27th July 2016 MCQSaumya SinhaОценок пока нет
- 7.MCQ 31st July 2016Документ5 страниц7.MCQ 31st July 2016Saumya SinhaОценок пока нет
- 29th July MCQДокумент5 страниц29th July MCQSaumya SinhaОценок пока нет
- MCQ August 2016Документ39 страницMCQ August 2016Saumya SinhaОценок пока нет
- Synthesis of Nico2o4Документ8 страницSynthesis of Nico2o4Amit kumarОценок пока нет
- Anodic and Cyclic VoltammetryДокумент30 страницAnodic and Cyclic Voltammetrysisipho sisipho100% (1)
- Nail Polish and Carbon Powder An Attractive Mixture To Prepare Paper Based ElectrodesДокумент7 страницNail Polish and Carbon Powder An Attractive Mixture To Prepare Paper Based ElectrodesMicu CristiОценок пока нет
- Analytical ChemistryДокумент7 страницAnalytical ChemistryNia RukmanОценок пока нет
- (EFC 39) Palmer, J.W. - Hedges, W. - Dawson, J.L. (Eds.) - Use of Corrosion Inhibitors in Oil and Gas Production-Maney Publishing (2004)Документ5 страниц(EFC 39) Palmer, J.W. - Hedges, W. - Dawson, J.L. (Eds.) - Use of Corrosion Inhibitors in Oil and Gas Production-Maney Publishing (2004)mirafrmОценок пока нет
- Corrosion and Surface Chemistry of Metals - Iran-Mavad - Com-205-409Документ205 страницCorrosion and Surface Chemistry of Metals - Iran-Mavad - Com-205-409Julio CastañedaОценок пока нет
- Acs Jpclett 2c01201Документ8 страницAcs Jpclett 2c01201NiladriОценок пока нет
- A New Route For The Electrodeposition of Platinum-Nickel AlДокумент6 страницA New Route For The Electrodeposition of Platinum-Nickel AlmomenziОценок пока нет
- Nano Energy: Xiang Wu, Shunyu YaoДокумент8 страницNano Energy: Xiang Wu, Shunyu YaoHamza QureshiОценок пока нет
- Flexible Glucose SensorДокумент7 страницFlexible Glucose SensorSAYYID HAMEEM N 20MTS0018Оценок пока нет
- 910 PSTAT Mini: Starting Out in The World of ElectrochemistryДокумент8 страниц910 PSTAT Mini: Starting Out in The World of ElectrochemistryabasakОценок пока нет
- 2 - Applications of Carbon Based Electrodes For Voltammetric Determination of Lornoxicam in Pharmaceutical Dosage Form and Human SerumДокумент11 страниц2 - Applications of Carbon Based Electrodes For Voltammetric Determination of Lornoxicam in Pharmaceutical Dosage Form and Human SerumhallyssonnnОценок пока нет
- PalmSens4 BrochureДокумент17 страницPalmSens4 BrochureKhalid ZghearОценок пока нет
- Admin,+3 +riyantoДокумент12 страницAdmin,+3 +riyantoAditya Rifqi PutraОценок пока нет
- TMB - Chronoamperometry-Based Redox Cycling For Application To ImmunoassaysДокумент7 страницTMB - Chronoamperometry-Based Redox Cycling For Application To ImmunoassaysLuciana FreireОценок пока нет
- Electroanalytical Chemistry A Series of AdvancesДокумент1 108 страницElectroanalytical Chemistry A Series of AdvancesFriscilla HermatasiaОценок пока нет
- POTENCIOSTATO Manual EC-Lab Software Techniques and ApplicationsДокумент269 страницPOTENCIOSTATO Manual EC-Lab Software Techniques and Applicationssalomonsan.rcОценок пока нет
- Cyclic TryДокумент12 страницCyclic TryfangmfengОценок пока нет
- Electrodeposition of Nanocrystalline Zn-Ni Coatings With Single Gamma Phase From An Alkaline BathДокумент10 страницElectrodeposition of Nanocrystalline Zn-Ni Coatings With Single Gamma Phase From An Alkaline BathIcanIbrahimОценок пока нет
- High Entropy Spinel-Structure Oxide For Electrochemical ApplicationДокумент8 страницHigh Entropy Spinel-Structure Oxide For Electrochemical ApplicationAlejadra ManotasОценок пока нет
- Review PecДокумент16 страницReview PecJuliana Herrera GuerraОценок пока нет
- PalmSens3: Portable Potentiostat/Galvanostat/EIS AnalyzerДокумент10 страницPalmSens3: Portable Potentiostat/Galvanostat/EIS AnalyzerAlejandro PereiraОценок пока нет
- Electrochemical Performance of NiFe2O4Документ8 страницElectrochemical Performance of NiFe2O4shehzad khanОценок пока нет
- Wu B 2014 PHD Thesis PDFДокумент269 страницWu B 2014 PHD Thesis PDFHéctor CortezОценок пока нет
- 4 Paper 4, J Coord ChemДокумент17 страниц4 Paper 4, J Coord ChemMuhammad IqbalОценок пока нет
- 1 s2.0 S0039914022006154 MainДокумент14 страниц1 s2.0 S0039914022006154 MainwardaninurindahОценок пока нет
- Journal of Alloys and Compounds: D.A.L. Almeida, A.B. Couto, N.G. FerreiraДокумент8 страницJournal of Alloys and Compounds: D.A.L. Almeida, A.B. Couto, N.G. FerreiraParom WaikasikarnОценок пока нет
- Lateral Layered Heterostructure Bimetallic Selenides Bi2Se3-FeSe2 @C ForДокумент10 страницLateral Layered Heterostructure Bimetallic Selenides Bi2Se3-FeSe2 @C ForSena KulaksızОценок пока нет
- Benzo PirenoДокумент6 страницBenzo PirenoAna Paula Mota FerreiraОценок пока нет
- Antalgin 2Документ11 страницAntalgin 2lindaОценок пока нет