Академический Документы
Профессиональный Документы
Культура Документы
Temperature Programmed Desorption With Reaction
Загружено:
aquacobiaАвторское право
Доступные форматы
Поделиться этим документом
Поделиться или встроить документ
Этот документ был вам полезен?
Это неприемлемый материал?
Пожаловаться на этот документАвторское право:
Доступные форматы
Temperature Programmed Desorption With Reaction
Загружено:
aquacobiaАвторское право:
Доступные форматы
0
Abstract
Temperature programmed desorption (TPD) with reaction is a useful technique for characterising catalysts.
A general model that simulates TPD with reaction experiments was developed and successfully validated.
The generality of the model means multiple catalytic sites may be specified, each having a distribution of
activation energy. Additionally, any reactant/catalyst system may be modelled by specifying the reactions
involved in the system. Three case studies were performed where the model was fit to experimental TPD
data from the literature. Some experiments were performed on one of the systems studied
(isopropylamine/ZSM-5) in order to better understand critical underlying processes. Quantitative
information such as distribution of activation energies for each catalytic site, pre-exponential factors and
relative site populations were extracted from the cases studied. The values obtained compare well with
those reported in the literature and with values determined using other quantitative techniques.
Temperature Programmed
Desorption with Reaction
Developing a general model for quantitative analysis
Karim Al Shebani Al Nehlawi
Homerton College
Partner: Qing (Chely) Miao Supervisor: Dr. Patrick Barrie
40 pages + Preface + Title Page + Safety Appendix
1
Contents
1. Introduction and aims ............................................................................................................................................... 2
1.1 What is temperature programmed desorption? .............................................................................................. 2
1.2 What are the benefits of TPD with reaction? ................................................................................................... 3
1.3 Aims of this research project ............................................................................................................................ 3
2. Literature review ....................................................................................................................................................... 4
2.1 Qualitative analysis ........................................................................................................................................... 4
2.2 Simple quantitative analysis ............................................................................................................................. 7
2.3 Quantitative analysis for determining kinetic parameters ............................................................................... 8
2.4 Brief description of isopropylamine/ZSM-5 system ....................................................................................... 11
3. Experiments ............................................................................................................................................................ 12
3.1 Experimental procedure ................................................................................................................................. 13
3.2 Thermal decomposition of propene ............................................................................................................... 14
3.3 Desorption of propene from ZSM-5 ................................................................................................................ 15
4. Model description ................................................................................................................................................... 16
4.1 Background knowledge ................................................................................................................................... 16
4.2 Generating a TPD curve .................................................................................................................................. 19
5. Model validation and discussion of results ............................................................................................................. 23
5.1 Comparison with specific model ..................................................................................................................... 24
5.2 Case Study: CO on nickel ................................................................................................................................. 25
5.3 Case Study: formic acid on copper .................................................................................................................. 30
5.4 Case Study: Isopropylamine on ZSM-5 ........................................................................................................... 34
6. Conclusion ............................................................................................................................................................... 37
7. Further work ........................................................................................................................................................... 37
8. Nomenclature ......................................................................................................................................................... 37
9. Works cited ............................................................................................................................................................. 38
10. Safety appendix ................................................................................................................................................... 41
2
1. Introduction and aims
Catalysts play a vital role in a wide variety of industries and understanding how they perform their function
can help in designing superior catalysts. Many financial, environmental and social benefits have arisen
from the study of catalysts, such as catalytic converters in car exhausts and catalytic crackers in oil
refineries. Characterising catalysts in order to extract functional information is a key step in developing a
thorough understanding of catalyst performance and can enable process improvements to be made.
1.1 What is temperature programmed desorption?
Temperature programmed desorption (TPD) is an experimental technique used to characterise catalysts
(King, 1975). It provides a wealth of information and is exclusive to the investigation of solid catalysts with
gaseous species. Typically, a catalyst sample is placed in a reactor with a heating element such that the
temperature can be programmed accordingly. The flow rate of gaseous species into the reactor can be
controlled and a mass spectrometer is usually used to detect the species leaving the reactor. The catalyst
may be subjected to pre-treatment as necessary, such as heating with an inert carrier gas to eliminate
adsorbed water vapour or bombarding with argon ions to clean a metal crystal surface. A gaseous probe
molecule is then injected into the reactor (usually in pulses of known volume) until the required coverage
has been attained on the catalyst. The remainder of the experiment can either be conducted with
continuous flow of a carrier gas in vacuum conditions. The temperature is then ramped linearly with time
so that:
o
) ( T t t T + = | where | is the heating rate (K/s) and
o
T is the initial temperature (K).
As the temperature increases, the rate of desorption from the catalyst initially increases and the coverage
on the catalyst decreases. However at some point in the experiment the coverage becomes small enough
that the rate of desorption decreases with temperature. There is therefore a maximum in the rate of
desorption at a particular temperature. A peak shape is thus characteristic of TPD results when plotted as
the desorption rate of species versus temperature. Figure 1 below depicts the set-up of a TPD experiment.
The catalyst sample could be a packed bed, a differential packed bed (very thin layer of catalyst) or an
exposed flat surface. Note that the concentration of adsorbed species is usually defined using a
dimensionless value of coverage, namely the ratio of occupied sites to total sites on a catalyst.
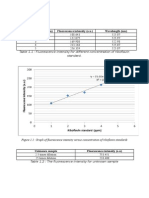
Figure 1: Typical set-up for a TPD experiment
Figure 2: TPD of ammonia on H-mordenite and ZSM-5 zeolites.
The mordenite curve has been vertically shifted to make it easier
to see (Niwa & Katada, 1997).
3
For illustration, Figure 2 above shows a TPD experiment using ammonia to probe the sites of H-mordenite
and ZSM-5 zeolites; note that ammonia desorbs without reaction in this case. The vertical axis is
proportional to the rate of desorption, and the horizontal axis could be interchanged between time and
temperature since they vary linearly with respect to each other. Peaks that occur at higher temperatures
indicate stronger binding energies to the sites on the catalyst; additionally, the area under the TPD curve is
proportional to the population of sites on the catalyst. In this case, the catalysts show two peaks, indicating
that there are two types of sites on which ammonia adsorbs, one strong and one weak. It is also clear that
mordenite contains more sites of higher binding strength than ZSM-5. This simple example shows how
useful TPD could be as a tool to characterise catalysts.
1.2 What are the benefits of TPD with reaction?
For the most part, species which do not undergo reaction on the catalyst are chosen for TPD studies so as
to simplify analysis. However, under certain circumstances, it may be beneficial to select a molecule that
does undergo reaction, resulting in different species desorbing from the catalyst; this is termed TPD with
reaction. TPD with reaction allows one to probe specific sites on a catalyst and can provide information on
reaction mechanisms and the activation energies for reaction. For instance (with reference to Figure 2
above), more information may be gained on ZSM-5 by using a reactive species such isopropylamine that is
believed to decompose only on strong Brnsted sites, rather than using ammonia. The products of the
reaction are ammonia and propene, which can be detected using the mass spectrometer, providing data
specific for characterising the Brnsted acid sites of a zeolite. However, detailed interpretation of results
becomes much more complicated when reaction occurs on the catalyst. There are many unknown
variables that make accurate quantitative data analysis difficult, to the extent that Falconer & Schwarz
(1983) remarked that a complete theoretical description of TPD with reaction (TPDR) is not likely. Despite
this, it might be possible to extract important information from TPDR with well designed experiments and
carefully chosen reactive molecules. However, thus far, the conclusions derived in the literature from TPD
with reaction studies have been predominantly qualitative.
1.3 Aims of this research project
The key aim of this research project is to develop a general model that can be used to extract quantitative
information from TPD with reaction experiments. The flexibility of a general model means that it could be
used on various reactant/catalyst systems with different reaction mechanisms at work. Consequently, the
next aim of this project is to validate the model by applying it to several case studies found in the literature
and comparing the results attained with those available in the literature. The final aim is to conduct TPD
4
with reaction experiments on the isopropylamine/ZSM-5 system and attempt to extract quantitative
parameters using the general model developed.
2. Literature review
This section reviews the types of analysis currently performed on TPD data and the information that can be
extracted as a result. The power and versatility of TPD experiments as a tool for characterising catalysts is
demonstrated and the difficulties associated with accurately determining quantitative parameters are
discussed, especially with reference to reactive systems. Furthermore, a brief description of the
isopropylamine on ZSM-5 system is given.
2.1 Qualitative analysis
The qualitative analysis that can be performed on TPD data is best illustrated by giving specific examples.
Peak Temperature & Area under TPD Curve
In general, a peak on a TPD curve represents desorption from a type of site on the catalyst surface.
However, bear in mind that peaks may overlap and it can become difficult to separate individual sites from
each other. The temperature at which the maximum rate is observed for a peak is called the peak
temperature, T
p
. Sites with stronger binding energies to the adsorbate will have higher values of T
p
and the
peaks will be shifted to higher temperatures. The area under a TPD curve is proportional to the amount of
adsorbate that has desorbed i.e. the area is proportional to the change in coverage of an adsorbate during
the TPD experiment. Accordingly, the area under a peak is proportional to the population of the site it
corresponds to, such that a more populous site would have a larger peak area. This information can be
used to compare and contrast different catalysts. Moreover, it can be used to evaluate how a catalyst
changes as a result of different preparation methods, composition, stages of deactivation or other
properties. The TPD curve is also a unique identifier for a catalyst and can be used for quality control
purposes when manufacturing large quantities of catalyst or indeed when preparing small batches in the
laboratory.
Varying Initial Coverage
TPD curves are also used extensively for determining catalytic reaction mechanisms and orders of reaction.
Take for example, the reaction of an adsorbate to produce a species that desorbs instantly, such that the
TPD peak observed for the product is indicative of the reaction and not the desorption of the product. If
the reaction is first order, then the value of the peak temperature T
p
does not change with initial coverage
of the adsorbed reactant; however, if the reaction is second order or higher, then T
p
will shift to lower
values as the initial coverage is increased. Therefore, one may perform several TPD experiments with
5
different initial coverages of the reactant to determine whether or not the reaction is first order.
Additionally, quantitative analysis can be performed to determine the order of the reaction; this is
discussed later. One problem is that the product species may readsorb onto catalyst downstream in the
reactor, particularly when porous catalysts are used. The effect this has is to delay the TPD curve and shift
it to higher temperatures (Gorte, 1982), which adds another degree of complexity that can hamper data
analysis. Another problem that arises is that the product does not desorb instantly and, as initial surface
coverage is increased, binding strength of the product usually decreases as a result of unfavourable
adsorbate-adsorbate interactions. This causes T
p
to shift to lower temperatures, potentially resulting in an
incorrect conclusion that the reaction is of order higher than one. Figure 3 below shows the TPD curves for
hydrogen desorption from a supported nickel catalyst at varying initial coverages. Molecular hydrogen
adsorbs dissociatively into two H
(ads)
atoms; therefore, the desorption process is second order because the
two atoms need to recombine and the effect this has on decreasing the peak temperature can be clearly
seen.
Figure 3: TPD of adsorbed hydrogen on a supported Ni catalyst.
Initial overage increases from (a) to (e) (Lee & Schwarz, 1982).
Figure 4: TPD of adsorbed CO on Ru(001) with varying initial
coverages. Note that the curves have been shifted vertically to
make it easier to see and the Langmuir L is a unit of coverage
(Brown & Vickerman, 1981).
Varying initial coverage can sometimes reveal more information. For example, Figure 4 shows the TPD of
CO on a surface crystal of Ru(001). As the initial coverage increases, a new peak labelled
2
appears. This
shows how adsorbed CO molecules preferentially adsorb onto the
3
site before the
2
site, suggesting the
3
site has higher binding strength. Moreover, this shows that the CO molecule is mobile on the surface of
Ru(001) since it can move to a
3
site if it were to first adsorb on a
2
site.
Varying Adsorption Temperature
The temperature at which molecules are adsorbed onto the catalyst can be varied in order to gain further
insight. If lower adsorption temperatures are used, this may reveal any weakly bound species. Higher
adsorption temperatures can eliminate weakly bound species from the TPD curve or promote reactions
that produce desired adsorbed species. For example, in order to determine whether surface carbon
6
species are involved in the mechanism of hydrogenation of CO on metal surfaces, CO can be adsorbed at
high temperatures on a metallic catalyst. This promotes the dissociation reaction that produces C
(ads)
and
O
(ads)
. A TPD experiment with hydrogen flowing as a carrier gas can then be performed at low
temperatures, with CO
(ads)
, C
(ads)
and O
(ads)
all present, which gives information on how adsorbed carbon
species take part in the reaction. For instance, this has been performed on a supported ruthenium catalyst
by Low & Bell (1979).
Using Isotopes
Mass spectrometers can differentiate between molecules containing different isotopes. Therefore, one
may mark specific atoms on a reactant using isotopes as a way of gaining more information on reaction
mechanisms. The mechanism for the Fischer-Tropsch reaction, which catalytically converts CO + H
2
into a
mixture of hydrocarbons and water, has been debated in the literature for many years. A popular
mechanism suggests CO
(ads)
dissociates into adsorbed C
(ads)
and O
(ads)
atoms, H
(ads)
atoms then combine with
C
(ads)
and O
(ads)
to form hydrocarbons and water. A key step in validating this mechanism is to prove that
CO
(ads)
actually dissociates on the catalyst and that it does so at a temperature similar to that at which the
Fischer-Tropsch reaction takes place. McCarty & Wise (1979) used isotope TPD methods that confirm the
dissociation of CO on a supported ruthenium catalyst.
12
C
18
O and
13
C
16
O were co-adsorbed in equimolar
amounts and a TPD experiment was performed, the results of which are shown in Figure 5 below.
Figure 5: TPD of
12
C
18
O and
13
C
16
O adsorbed on a alumina-supported Ru catalyst, indicating that reversible dissociation occurs.
The species
12
C
16
O and
13
C
18
O were desorbing in significant amounts at temperatures above 375 K,
indicating that reversible dissociation must be occurring. McCarty & Wise (1979) also quantitatively
determined that the exchange rate between isotopes peaked at 427 K, whereas the production of
methane during hydrogenation of adsorbed CO on the same catalyst peaked at 470 K. This shows that CO
dissociation occurs at lower temperatures than the Fischer-Tropsch reaction, supporting the reaction
mechanism discussed above.
7
2.2 Simple quantitative analysis
Quantitative data normally extracted from TPD data include (Falconer & Schwarz, 1983):
- Active surface area of catalyst
- Coverage of adsorbates
- Relative population of different sites
- Order of reaction
- Activation energy of desorption or reaction
- Pre-exponential factor for the rate constant
Active Surface Area of Catalyst
The area under a TPD curve is proportional to the number of molecules that have desorbed from the
catalyst. If an adsorbate achieved full coverage on a catalyst, then by calculating the number of molecules
that desorbed after a TPD experiment (assuming all molecules desorb) and by knowing the surface area
occupied by each molecule, the active surface area of the catalyst can be determined. Choosing an
adsorbate that does that not adsorb into multiple layers on the catalyst can simplify the analysis
significantly. The rate of desorption measured by the mass spectrometer is normally in arbitrary units;
therefore, a calibration experiment must be performed whereby a known amount of adsorbate is loaded
onto the catalyst and a TPD experiment is performed. The area under the resulting TPD curve is calculated,
which allows us to relate the arbitrary units of the mass spectrometer to known units of desorption, such
as mol/s. Attention must be paid to the fact that the sensitivity of some mass spectrometers varies with
different molecules such that it is often preferred to conduct this calibration for each molecule detected.
Coverage
Equation 1 below may be used to calculate the coverage u as a function of temperature T if the catalyst
was initially loaded with coverage of one monolayer (i.e.
o
u = 1). Note that this may not apply when
reactions are present that can produce other adsorbed species. Equation 2 may be used when the initial
coverage is not unity, but it requires the area for desorption of a monolayer to be known. The rate of
desorption is r.
( )
|
|
.
|
\
|
=
} }
f
o
o
T
T
T
T
dT
r
dT
r
T
| |
u 1
(1)
( )
} } }
=
|
|
.
|
\
|
=
f
o o o
T
f
o
T
T
T
T
T
dT
r
dT
r
dT
r
T
1 u
| | |
u (2)
Note ( )( ) dT d dt d r u | u = = .
8
Relative population of Sites
The area beneath a peak is also proportional to the population of its corresponding site. Therefore, if peaks
can be separated from each other, then the relative population of different types of site can be
determined; however, this becomes difficult when peaks overlap
2.3 Quantitative analysis for determining kinetic parameters
The kinetic parameters that are usually determined are the order of desorption/reaction, the activation
energy for desorption/reaction and the pre-exponential factor used for the rate constant. Numerous
methods have been devised for extracting these parameters, ranging from relatively simple ways to more
accurate yet complicated ways. Jong & Niemantsverdriet (1990) studied a wide sample of analysis methods
and concluded that simple, rough procedures yield unreliable results, but are easy to apply. Furthermore,
they suggest that complete methods, which make fewer assumptions and use several TPD curves to
extract parameters, should be used whenever possible, as they give better results. Each method is
discussed separately below.
Redheads Method
This is the simplest and most commonly used method for determining kinetic parameters. In a lot of cases,
especially with regards to reactive systems, several parameters that are needed as input for more
complicated methods are not known. Therefore, Redheads (1962) method offers a simple and easy to use
procedure that gives approximate results. For a system with no readsorption, the rate of desorption is
equal to the flow rate of desorbed molecules detected by the mass spectrometer. This is not the case with
readsorption because the total flow rate of species leaving the reactor is reduced. In the absence of
readsorption, the normalised rate detected by the mass spectrometer r is given by Equation 3 below. This
assumes a desorption process or a rate limiting reaction of order n in coverage u . At the peak temperature
T
p
the rate of desorption is a maximum implying that
2 2
0 dT d dt dr u = = . This condition produces the
relationship given by Equation 4 below, which can be used to calculate E
act
if the pre-exponential factor A is
known (or vice versa). The peak temperature and coverage at the peak temperature must also be known;
however, the coverage term cancels out when the order of reaction/desorption is one. The main problem
with Redheads method is that the pre-exponential factor is often not known and has to be estimated or
determined using statistical mechanics.
n n
RT
E
A k
dT
d
dt
d
r u u
u
|
u
|
.
|
\
|
= = = =
act
exp (3)
|
|
.
|
\
|
=
p
act
1
p
2
p
act
exp
RT
E
An
RT
E
n
|
u
(4)
9
Peak Width and Shape Index Methods
The following two methods of quantitative analysis are based around taking two or three points of data
from a single TPD curve and performing brief calculations to determine some kinetic parameters.
Peak width analysis utilises the width of a single TPD peak at half or at three-quarters of the maximum rate
of desorption (W
1/2
and W
3/4
), in addition to the peak temperature T
p
, to determine the activation energy
E
act
and the pre-exponential factor A. Falconer & Schwarz (1983) and Jong & Niemantsverdriet (1990) have
summarised and rearranged the equations used for this method in situations of first-order and
second-order kinetics into an easier format than those initially developed by Chan et al. (1978). These are
shown in Table 1 below. W
1/2
, W
3/4
, and T
p
do not vary with coverage for first-order situations, but do vary
for second-order. Therefore, the values of E
act
and A determined for second-order situations are a function
of coverage.
Table 1: Equations used for Peak Width Analysis.
o
is the inital coverage (Falconer & Schwarz, 1983; Jong & Niemantsverdriet, 1990).
First Order Second Order
|
|
.
|
\
|
+ + =
5 . 0
2
2 1
2
p
p act
832 . 5
1 1
W
T
RT E
|
|
.
|
\
|
+ + =
5 . 0
2
2 1
2
p
p act
117 . 3
1 1 2
W
T
RT E
|
|
.
|
\
|
+ + =
5 . 0
2
4 3
2
p
p act
353 . 2
1 1
W
T
RT E
|
|
.
|
\
|
+ + =
5 . 0
2
4 3
2
p
p act
209 . 1
1 1 2
W
T
RT E
|
|
.
|
\
|
=
p
act
p
act
exp
RT
E
RT
E
A
|
( )
|
|
.
|
\
|
+
=
p
act
p act
3 2
o
2
act
exp
2
p
RT
E
RT E T R
E
A
u
|
Another method is based upon calculating a shape index, which is the ratio of slopes at inflection points of
a single TPD curve, as shown in Equation 5 below. Ibok & Ollis (1980) concluded that this method allows
one to distinguish between systems that are first or second-order and to determine the extent of
readsorption. Both Ibok & Ollis (1980) and Criado et al. (1982) showed that when RT E
act
is large, the
value of the shape index for first and second-order kinetics without readsorption is independent of initial
coverage
o
u
.
However, Criado et al. (1982) discovered that the values of the shape index for first-order
systems without readsorption and second-order systems with readsorption may be similar and difficult to
distinguish. Table 2 below shows the values the shape index can take under different situations (Falconer &
Schwarz, 1983).
2
2
1
2
2
2
1
2
2
2
2
2
T
T
T
T
dT
d
dT
d
dT
r d
dT
r d
Index Shape
u
u
= = (5)
Note: T
1
and T
2
are the low and high inflection point
temperatures, respectively.
Table 2: Values of the shape index when the initial coverage
o
= 1, T
2
/T
1
1.1 (typical value) and E
act
/RT is large.
Without
Readsorption
With
Readsorption
First-Order 0.76 0.55
Second-Order 1.46 0.86
10
Varying the Heating Rate
TPD peak temperatures increase with the heating rate | . The problem of not knowing the pre-exponential
factor in Redheads method may be overcome by conducting several TPD experiments at different heating
rates, giving a series of TPD curves with different peak temperatures. Rearranging Equation 4 above results
in Equation 6 below; this shows that a plot of ( )
2
p
ln T | vs.
p
1 T results in a straight line of slope R E
act
.
This method assumes that for reactions of order greater than 1, the coverage at peak temperature
p
u is
constant, which is valid for second-order systems because the TPD curve is symmetrical in these cases.
p
u
is calculated from Equation 1 or 2. The pre-exponential factor can then be determined by the intercept of
the plot generated.
( )
p
act
1
p 2
p
ln ln
RT
E
R An
T
n
=
|
|
.
|
\
|
u
|
(6)
Varying the Initial Coverage
A series of TPD experiments can also be conducted at different initial coverages. Rearranging Equation 3
above gives Equation 7 below; this shows that a plot of ( ) r ln vs. ( ) u ln at constant temperature gives a
straight line whose slope is equal to the order of reaction n. The value of u at a particular temperature can
be retrieved by using Equation 2 above and the rate r can be read directly off the TPD curve. From a series
of TPD curves at various initial coverages, one may determine several data points (r, u ) at the same
temperature. However, when
act
E is a function of coverage (due to adsorbate-adsorbate interactions) then
the apparent order of reaction will vary with coverage. Falconer & Madix (1977) showed that if the
activation energy decreased with coverage (as is expected for adsorbates that repel each other), then the
apparent order of reaction increases. According to Falconer & Madix (1977), this is an easy and useful
method for determining whether or not the activation energy is a function of coverage. Furthermore, by
plotting ( ) r ln vs. T 1 at constant coverage then the activation energy can be determined from the slope,
and this allows one to derive the dependence of activation energy on coverage. Falconer & Schwarz (1983)
commented that this technique is not appropriate for TPD curves that show significant desorption from
multiple sites.
( ) ( ) ( ) u ln ln ln
act
n
RT
E
A r +
)
`
= (7)
Readsorption Considerations
Readsorption is when a desorbed species readsorbs onto catalyst further down the reactor. Gorte (1982)
has shown that it is very difficult to eliminate readsorption even when high carrier gas flow rates are used.
(Gorte, 1982) went further to say that analysing TPD data must be done with care and that in many
11
scenarios only qualitative results can be deduced. It is commonly found that the overall activation energy
corresponds to the heat of adsorption in this case, rather than the activation energy for desorption. These
parameters will be numerically the same if the adsorption process is non-activated (which is the case for
many systems of interest) (Gorte, 1982).
Non-Constant Activation Energy Considerations
Most of the above methods consider activation energy to be constant; however, in reality E
act
can vary with
coverage; additionally, there can be a distribution of activation energies for each type of site on a catalyst.
For example, this could be due to small environmental variations around each site. Such complicating
factors require numerical simulation of a TPD curve, followed by regressional fitting of the model to
experimental TPD data. This type of analysis has been performed less frequently in the literature;
nevertheless, it can be useful to measure the dependence of E
act
on coverage or to determine the
distribution of activation energies for a catalytic site. More importantly, this method utilises all the
information in a TPD curve, rather than just using a few data points, and so gives more confidence in the
quantitative results obtained.
Other methods, discussed previously in this section, have difficulty extracting quantitative parameters
from overlapping TPD peaks because the theory behind each method assumes desorption/reaction from a
single site. However, experimental TPD curves are very often a convolution of several overlapping peaks.
This requires analysis by computational methods with regressional curve fitting to model several sites on a
catalyst simultaneously. Moreover, this enables the relative population of different types of site on a
catalyst to be quantified.
This project will focus on numerical modelling of TPD with reaction by assuming there is a distribution of
activation energies for each site. It will be demonstrated later this method can prove very useful for
extracting reasonable quantitative results from complicated and overlapping TPD curves. Furthermore, it is
useful for systems undergoing complicated reaction mechanisms.
2.4 Brief description of isopropylamine/ZSM-5 system
Isopropylamine is basic in nature and can adsorb onto acid sites within zeolites such as ZSM-5. The
structure of isopropylamine is shown in Figure 6 below. If a TPD experiment is performed on a sample of
ZSM-5 that has been fully covered with isopropylamine, then at temperatures below 500 K some weakly
bound isopropylamine desorbs unreacted; however, at temperatures between 550 K and 650 K,
isopropylamine reacts to form ammonia and propene (Parrillo, et al., 1990; Miao, 2012). The overall
reaction is shown below. Our goal is to extract quantitative kinetic parameters relevant to the
12
isopropylamine/ZSM-5 system. It is believed that the reaction to form propene and ammonia might be rate
limiting because ammonia and propene are reported to desorb at the same temperature in TPD
experiments (Parrillo, et al., 1993). However, some issues have been raised regarding this system, including
the fact that propene is known to undergo reaction at high temperatures which may complicate the
modelling process. Also, the reaction step may not be entirely rate limiting; desorption of propene and
ammonia may potentially be slow enough that the TPD peaks observed are dependent on desorption
rather than the reaction kinetics. Finally, readsorption in a porous catalyst such as ZSM-5 is likely and this
will complicate the analysis.
H
3
C
NH
2
H
C
CH
3
Figure 6: Structure of isopropylamine.
3 2 3 3 2 3
NH CHCH CH )CH CH(NH CH +
Parrillo et al. (1990) discovered that the amount of isopropylamine that reacted in ZSM-5 corresponded to
a 1:1 ratio with aluminium atoms present. Brnsted sites are formed in ZSM-5 when an Al and a Si atom
are linked by a bridging OH hydroxyl group on the surface of a pore. As a result, Parrillo et al. (1990)
concluded that isopropylamine only reacts on strong Brnsted acid sites, with one isopropylamine
molecule adsorbing per Brnsted site. This is useful because the TPD with reaction experiment with
isopropylamine is thus giving information specific to Brnsted acid sites, rather than any other sites that
may be present. Furthermore, Parrillo & Gorte (1992) showed that the heat of adsorption of
isopropylamine on ZSM-5 is constant at 240 kJ/mol, up to a coverage of one molecule per Al atom. As a
result, they concluded that all Brnsted acid sites in ZSM-5 are of equal strength, which is also supported
by Kresnawahjuesa et al. (2002) and by the results of this report (discussed later). Assuming adsorption is
not activated, the heat of adsorption will equal the activation energy for desorption. Using Redheads
method with assuming A = 10
13
s
-1
(the expected value for a unimolecular desorption process)
(Barrie, 2012), an activation energy of 240 kJ/mol results in a peak temperature of about 870 K for a
heating rate of 1 K/s. This explains why the isopropylamine on Brnsted sites are observed to react before
desorbing because the temperature at which desorption is expected (870 K) is much higher than that at
which the reaction occurs (550-650 K).
3. Experiments
My role within the project was mainly in developing and validating a general model for TPD with reaction;
however, during the first half of the project, I conducted some experiments in collaboration with my
partner, Qing (Chely) Miao. These experiments will be discussed in the following two sections, but due to a
13
lack of space, only experiments that produced useful data will be covered. My partner and I performed
many more experiments that yielded unreliable data due to difficulties experienced with the experimental
set-up (especially with regards to the isopropylamine experiments). My partner later performed more
experiments on the isopropylamine/ZSM-5 system on her own, the details of which may be found in her
report.
The experiments described in this section aim to answer the following questions addressed in section 3.4:
- Does propene react to form other species within the temperature range relevant for the
isopropylamine/ZSM-5 system?
- At what temperature does desorption of propene from ZSM-5 occur? Is it rate limiting in the
temperature range of the isopropylamine/ZSM-5 reaction?
Unfortunately, we could not perform experiments with ammonia because it was unavailable. Moreover,
issues regarding safety prevented my partner from using ammonia in further experiments.
3.1 Experimental procedure
The apparatus used consists of an integrated microreactor and quadrupole mass spectrometer (see
Figure 1), manufactured by Hiden CATLAB Systems. Up to four gas cylinders could be connected, whereby
the flow rate of different gases entering the reactor could be individually controlled. The reactor used
consists of a hollow quartz tube that is able to withstand high temperatures. The tube was cleaned using
hexane, isopropanol and acetone (in that order) and allowed to dry completely before use. Quartz wool
was inserted in the tube before and after the catalyst sample to ensure the catalyst remained in place and
to prevent any particulates from entering the mass spectrometer. A heating element surrounding the tube
facilitated programming the temperature automatically with feedback from a thermocouple placed inside
the tube. The temperature could be controlled from 323 1173 K. A split-stream from the reactor is sent
to the mass spectrometer for analysis.
The first experiment conducted aimed to determine the temperature range over which propene undergoes
thermal decomposition. No catalyst was used for this experiment. A continuous flow of propene (2 ml/min)
and helium (38 ml/min) were passed through the reactor tube while the temperature was ramped from
323 K to 1148 K at a rate of 2 K/min. The mass spectrometer was set to scan mass-to-charge (m/e) ratios
1 42. This allowed us to determine the main products of reaction. The second experiment aimed to
perform TPD on a sample of ZSM-5 with full initial coverage of propene. The ZSM-5 sample used weighed
25 mg. ZSM-5 adsorbs water vapour from air so samples were pre-treated by heating at 873 K for about
one hour in a continuous flow of helium. The mass spectrometer was set to detect water leaving the
reactor so as to ensure all water was eliminated at the end of pre-treatment. The reactor was then cooled
14
down to 323 K while maintaining the flow of helium over the catalyst. Propene was then injected into the
helium carrier gas by using small pulses (volume = 250 l at atmospheric pressure) while the concentration
of propene leaving the reactor was detected. Pulsing continued until the peak height of pulses, on a plot of
concentration versus time, came to a steady height. This indicates that the catalyst has been fully loaded
with propene (11 pulses were required). The temperature was then ramped from 323 1148 K at a
constant rate of 5 K/min. The helium carrier gas flow rate of 40 ml/min was maintained, while the mass
spectrometer was set to detect m/e = 41 (corresponding to the main peak of propenes spectrum).
Sections 3.2 and 3.3 below present the experimental results my partner and I obtained from the
experiments described above. The results are also discussed and subjected to a critical analysis.
3.2 Thermal decomposition of propene
The results obtained from the experiment to determine when thermal decomposition of propene occurs is
shown in Figure 7 below. As can be seen, propene passes unreacted until about 1050 K where the
concentration of propene (m/e = 41) drops suddenly and the concentration of other species increases.
Unfortunately, the experiment could not be continued at higher temperatures to see the extent of reaction
because of equipment limitations. The m/e ratios shown in Figure 7 were selected because they were the
only ones that showed significant change upon reaction. The m/e ratios of 2, 15, 16, 26 and 28 were seen
to increase once decomposition occurred. These have been attributed to hydrogen, methane and ethene,
as shown in Table 3 below. An extremely minute increase in the m/e = 30 was also detected, indicating
very small quantities of ethane were being produced as well. It is relevant to note that some propene ions
fragment in the mass spectrometer to give signals at m/e = 15 and m/e = 26. Laidler & Wojciechowski
(1960) reported the same major products for propene decomposition. The temperature range over which
propene was detected in my partners isopropylamine/ZSM-5 TPD experiment is 550 660 K (Miao, 2012).
Therefore, this indicates that the thermal decomposition of propene will not be a complicating issue in
modelling this system.
Figure 7: Continuous flow of propene with a constant heating
rate. Decomposition occurs at about 1050 K. m/e ratios of 2, 15,
16, 26 and 28 are seen to increase upon reaction.
Table 3: A tick indicates the m/e value is present
in the spectrum. A double tick indicates the m/e
value is the major peak. An indicates the m/e
value is not present in the spectrum (NIST, 2011).
m/e Propene Ethene Methane Hydrogen
2
15
16
26
28
41
0.00E+00
5.00E-09
1.00E-08
1.50E-08
2.00E-08
2.50E-08
3.00E-08
3.50E-08
4.00E-08
350 550 750 950 1150
M
a
s
s
S
p
e
c
S
i
g
n
a
l
(
a
u
)
Temperature (K)
m/e = 41
m/e = 28
m/e = 26
m/e = 16
m/e = 15
m/e = 2
15
3.3 Desorption of propene from ZSM-5
The result of our TPD experiment involving propene on ZSM-5 is shown in Figure 8 below. The coverage of
propene at each temperature has been calculated using Equation 1 and is shown in Figure 9 below.
Figure 8: TPD of propene on ZSM-5 with full initial coverage.
Figure 9: Calculated coverage of propene on ZSM-5 during TPD.
Figure 8 shows that propene mainly desorbs between 360 K and 650 K. Three overlapping peaks can be
distinguished, indicating at least three sites of different binding energies. The peak temperature of the site
with the highest binding energy is about 607 K. The reaction of isopropylamine on ZSM-5 occurs between
550 K and 660 K, with a peak temperature of about 630 K. Therefore, propene desorption is quicker than
isopropylamine reaction at all temperatures. Using Redheads method for a first order process (assuming
A = 10
13
s
-1
, | = 1 K/s), peak temperatures of 607 K and 630 K result in activation energies of 166 kJ/mol
and 172 kJ/mol. Using Equation 3, the ratio of the rate of desorption to the rate of reaction at unity
coverage has been calculated and is shown in Figure 10 below. As can be seen, the rate of propene
desorption is about four times as large as the rate of reaction between 550 K and 660 K. This indicates that
a peak temperature difference of 23 K is large enough that desorption will not limit the progress of the
reaction. It also means that propene can be considered to desorb instantly once produced by the reaction
for the purposes of modelling. On a separate note, coke was found to form in the ZSM-5 catalyst during the
experiment. This indicates that propene undergoes reaction on ZSM-5; however, it is believed that only
trace amounts of coke formed and thus the extent of reaction is small.
Figure 10: The ratio of the rate of desorption to the rate of reaction at full coverage has been calculated using Equation 3.
0.E+00
1.E-10
2.E-10
3.E-10
4.E-10
5.E-10
300 500 700 900 1100
M
a
s
s
S
p
e
c
S
i
g
n
a
l
(
a
u
)
Temperature (K)
0.0
0.2
0.4
0.6
0.8
1.0
300 500 700 900 1100
C
a
l
c
u
l
a
t
e
d
C
o
v
e
r
a
g
e
Temperature (K)
0
1
2
3
4
5
6
500 550 600 650 700
Temperature (K)
R
a
t
i
o
o
f
D
e
s
o
r
p
t
i
o
n
t
o
R
e
a
c
t
i
o
n
R
a
t
e
s
16
4. Model description
The following section will describe the model which I developed with guidance from my supervisor
Dr. Patrick Barrie. My partner did not contribute to the development of this model.
The model developed uses fundamental rate laws to fit computationally calculated TPD curves to
experimental data. As a result, quantitative parameters may be determined such as distribution of
activation energies for sites, pre-exponential factors and the relative population of different sites on a
catalyst. Another useful characteristic of the model is the fact that it is general, rather than being specific
to a particular reactant/catalyst system. This allows a wide range of systems to be analysed with relative
ease and practicality, as opposed to individually deriving and programming different sets of differential
equations for each system. The model can also be used on systems not undergoing reaction, as will be seen
shortly.
Quantitative analysis of TPD with reaction data is challenging because of the complexity of some reactive
systems and the presence of readsorption. The model developed enables extraction of meaningful kinetic
parameters from complex reacting systems; however, due to time restrictions, readsorption has not been
accounted for in the model. MATLAB was used as the programming environment because of its utilisation
of matrix variables that make manipulation of large quantities of data more efficient.
4.1 Background knowledge
The details of the programming code will not be covered in this report, but have been deposited with my
supervisor. In order to define the equations forming the foundation of the model, the following will review
some necessary background knowledge.
Reaction set:
The simplest way to explain how a set of reactions is input into the model is by way of example. Let us
consider we have a molecule
(ads)
A that has been adsorbed on to a catalyst.
(ads)
A undergoes simple
desorption to form
(g)
A without reaction.
(g)
A is then carried away quickly enough such that readsorption
does not occur. Therefore, in this system the reaction set consists of one reaction, shown in Reaction Set
1 below:
(g) (ads)
A A
Reaction Set 1
(g) (ads) (ads) (g)
2B 2B A A
Reaction Set 2
Note that even though only desorption and no reaction is occurring, for the purposes of the model,
(ads)
A
and
(g)
A can be considered to be two separate species with a non-reversible reaction taking place between
them. Reaction Set 1 thus consists of two species and one reaction. Therefore, within the model, species
are differentiated not only by their atomic makeup, but also by their physical state (i.e. adsorbed or
17
gaseous). Let us consider another example, where this time molecule
(ads)
A can also undergo a reversible
surface reaction to form two molecules of
(ads)
B that later desorb non-reversibly to give two molecules of
(g)
B ; this scenario is illustrated in Reaction Set 2 above. This situation can be modelled using four species
(ads)
A ,
(g)
A ,
(ads)
B and
(g)
B , and the reaction set will consist of four reactions, namely:
(g) (ads)
A A ,
(ads) (ads)
2B A ,
(ads) (ads)
A 2B and
(g) (ads)
B B .
The stoichiometric matrix S stores the stoichiometric coefficients for each species in each reaction taking
place. Therefore, the stoichiometric matrix for Reaction Set 1 above is | | 1 1 = S , where the -1 represents
(ads)
A being consumed and the 1 represents
(g)
A being produced. Similarly, the stoichiometric matrix for
Reaction Set 2 above is shown below. The first row represents
(g) (ads)
A A , the second row
(ads) (ads)
2B A ,
the third row
(ads) (ads)
A 2B and finally the fourth row represents
(g) (ads)
B B .
(
(
(
(
=
1 1 0 0
0 2 1 0
0 2 1 0
0 0 1 1
S
The physical state of species matrix, isAdsorbed, stores information on whether each species is adsorbed or
gaseous. For the species involved in Reaction Set 2 above, | | 0 1 1 0 = isadsorbed where a 0 means the
molecule is not adsorbed and a 1 means the molecule is adsorbed. This feature is required because if the
products of a surface reaction occupy more sites than the reactants, such as
(ads) (ads)
2B A , then the rate
of reaction is given by ( )
B A A
1 u u u = k r , where ( )
B A
1 u u is the coverage of empty sites on the
catalyst. As a result, the model must know which molecules are adsorbed and which are in the gaseous
state in order to calculate
sites empty
u . Therefore, to define a reaction set, one must input four pieces of
information into the model:
- The number of species being modelled, which is called numOfMolecules.
- The number of reactions being modelled, which is called numOfReactions.
- The stoichiometric matrix S, which is of size (numOfReactions x numOfMolecules).
- The physical state of molecules matrix called isAdsorbed, which is of size (1 x numOfMolecules).
Number of sites and distribution of activation energy for each site:
In order to reproduce TPD curves with multiple sites, such as that shown previously in Figure 4, the model
needs to be able to simultaneously compute reaction/desorption from multiple sites. The relative
population of each type of site will thus need to be considered. For example, site type 1 may make up 30%
and site type 2 may make up 70% of the catalyst. This information does not need to be known beforehand
18
because it is considered a variable than can be changed in order to fit experimental data. In this way, the
relative population of different sites is one of the quantitative results produced by the model.
Every reaction being modelled, be it in reality desorption or an actual reaction, has associated with it an
activation energy. Most quantitative techniques found in the literature assume there is a single activation
energy for each site; however, this is not always the case, and this model considers a normal distribution of
activation energy for each type of site. Not only is this more realistic, but it also allows the model to
produce a better fit to experimental TPD data and gives an idea of how wide is the distribution of
activation energies is for each type of site. This may potentially provide critical information about the
environment in which adsorbed species are present and the reaction mechanisms at work.
The normal distribution of activation energy for each type of site is accounted for by considering every
type of site to consist of numerous points, each of which have a single activation energy. The activation
energy of the points will be spread across the distribution of activation energy for the site type. For
example, if we have one type of site with a normal distribution of activation energy, then we can model
that distribution by considering the site type to consist of say 50 point sites, each of constant activation
energy. These smaller sites or points will have activation energies between o 3
mean
E and o 3
mean
+ E ,
where
mean
E and o are the mean activation energy and the standard deviation for the site type. Figure 11
below illustrates the difference between a site type and a site point. Each point can be considered to be a
stand-alone site when modelling, so if there are two types of site, each of which having a distribution
modelled using 50 points, then effectively we can consider there to be 2x50 = 100 sites present. However,
the relative population of the different points being used for each site distribution needs to be considered.
A matrix called will store the fractional weighting every point has for each site distribution. For example,
a point with activation energy equal to
mean
E will have a larger fractional weighting than a point with
activation energy o 3
mean
+ E , because there are more sites with the mean activation energy than with an
activation energy on the outskirts of the distribution.
Figure 11: The difference between a site type and a site point.
19
Finally, the pre-exponential factor for the rate constant in each reaction must be defined. This is assumed
not to vary with temperature or between sites because it is intrinsic to the molecules taking part in the
reaction/desorption. In reality the pre-exponential factor may in fact vary with temperature (Miller, et al.,
1987); however, the assumption that it is constant is commonly made by most analytical methods to allow
feasible progress in quantifying kinetic parameters. So in summary the following parameters are in use:
- The number of sites, called numOfSites.
- The fractional population of each site matrix, called of size (1 x numOfSites).
- The mean activation energy matrix called
mean
E which is of size (numOfReactions x numOfSites).
- The standard deviation of activation energy matrix called which is of size
(numOfReactions x numOfSites).
- The number of points to consider for each site, called numOfPoints.
- The fractional weighting matrix called which is of size
(numOfReactions x numOfSites x numOfPoints).
- The pre-exponential factor matrix A which is of size (1 x numOfReactions).
Note that the model allows the user to define a different activation energy distribution for every reaction
on each site. However, it is assumed that the activation energy is perfectly correlated to the site binding
strength; as such, the distributions of activation energy for all reactions on a particular site will have the
same standard deviation. This assumption has been made through valid physical insight in order to simplify
computation.
4.2 Generating a TPD curve
TPD with Simple Desorption:
Consider
(ads)
A , of coverage u , undergoing desorption to produce
(g)
A without readsorption. The governing
rate equation is shown below.
n
RT
E A
dT
d
u
|
u
|
.
|
\
|
=
act
exp (8)
Our aim is to produce a graph of dT du versus T . In order to do that, u must first be calculated for all
temperatures. Assuming no distribution of activation energy, this is done by numerically integrating
Equation 8 above to yield the coverage as a function of temperature, ( ) T u , which is later plugged back into
Equation 8 to calculate dT du as a function of temperature. We have chosen to plot dT du
rather
than dt du
versus T so that area beneath the curve equals initial coverage. This eases analysis and
comparison with experimental data, as shown later.
20
The situation changes when there is a distribution of activation energy. Recall that each site point could
be considered as a stand-alone site for the purposes of modelling. Thus, we need to distinguish between
i site
u , which is the overall coverage of site type i, and between
j i point , site
u , which is the coverage of site
point j corresponding to site type i, i.e. the population of occupied site point j divided by the total
population of site point j. Equation 8 above will now apply to each site point, rather than applying to the
entire site type, and the coverage as a function of temperature for each site point will have to be
determined. The rate of desorption from all site points are then summed up to yield the overall rate of
desorption from site type i, see Equations 9 and 10 below. The population weighting of every site point, is
stored in matrixc , whereby the element
ij
c equals (population of site point j corresponding to site type i) /
(population of all site points corresponding to site type i).
ij
E is the activation energy of site point j
corresponding to site type i.
=
)
`
=
s numOfPoint
j
j i
ij
i
dT
d
dT
d
1
point , site site
u
c
u
(9)
=
)
|
|
.
|
\
|
=
s numOfPoint
j
ij
ij
i
RT
E
A
dT
d
1
site
exp
|
c
u
(10)
Extra consideration is needed when more than one site type is present. The overall rate of desorption
dT du , is given by Equation 11 below. The population fraction for site type i is given by
i
o , which is a
number between 0 and 1, such that if site type i makes up 30% of all sites on the catalyst, then 30 . 0 =
i
o .
= =
)
|
|
.
|
\
|
=
numOfSites
i
s numOfPoint
j
j i
ij i
dT
d
dT
d
1 1
point , site
u
c o
u
(11)
Therefore, Equation 8 must be solved to give the coverage of each site point, which then allows
dT d
j i site point ,
u to be calculated for each site point. This in turn is used in Equation 11 to yield the
desorption rate of A molecules as function of temperature.
Example TPD Simulation: Effect of varying E
mean
, and :
Consider the desorption
(g) (ads)
A A from a catalyst with full initial coverage of
(ads)
A . There is only one
type of site on the catalyst, which has a normal distribution of activation energy for desorption of
(ads)
A .
The base case considered has the following parameters:
mean
E
= 100 kJ/mol, = 10 kJ/mol, A = 10
13
s
-1
,
|
= 1 K/s. Figures 12, 13 and 14 below are plots of desorption rate (K
-1
) versus temperature (K). They show
the effect of changing
mean
E , and | , respectively. Note, that the y-axis in these figures is called
Desorption Rate of A, which refers to dT du rather than dt du . The area under each curve was
verified to be 1 for all cases considered below.
21
Figure 12: The effect of changing E
mean
, with = 10 kJ/mol and
= 1 K/s.
Figure 13: The effect of changing , with E
mean
= 100 kJ/mol and
= 1 K/s.
Figure 14: The effect of changing , with E
mean
= 100 kJ/mol and = 10 kJ/mol.
Digression: Defining the units for amount of gaseous species:
We need to incorporate the presence and reaction of multiple species, both in adsorbed and gaseous
states. The concentration of adsorbed species is defined using the dimensionless value of coverage, which
is the ratio of occupied sites to total sites on a catalyst, as explained before. Since the total number of sites
on a catalyst remains constant, this means coverage is directly proportional to the amount of adsorbed
species. Recall that this model does not include readsorption; therefore, the concentration of a species in
the gas phase is never included in any rate equation because it is assumed that gaseous species are
transported away quickly enough not to readsorb. Consequently, the model will only keep track of the
amount of each gaseous species that has desorbed from the catalyst and the rate at which gaseous species
being produced, not the concentration. The unit of gaseous amount used is defined such that if a
monolayer of coverage ( ) 1 = u desorbs completely, this will correspond to a gaseous amount of 1. This is
done for convenience and for simplifying comparison of the amount of adsorbed species in terms of
coverage to the amount of gaseous species desorbed. Also, in this way, the rate of desorption of an
adsorbed species will equal the rate of production of its corresponding gaseous species - had we used
moles for the amount of gaseous species, a constant of proportionality would be required to equate these
two rates. Moreover, to simplify the equations involved (and to simply the code underlying the model), the
nomenclature for gaseous amount used will also be u , such that for the specific case of
(g) (ads)
A A , the
0.000
0.002
0.004
0.006
0.008
0.010
0.012
200 300 400 500
N
o
r
m
a
l
i
s
e
d
D
e
s
o
r
p
t
i
o
n
R
a
t
e
(
K
-
1
)
Temperature (K)
Emean = 90 kJ/mol
Emean = 100 kJ/mol
Emean = 110 kJ/mol
0.000
0.004
0.008
0.012
0.016
0.020
200 250 300 350 400 450 500
N
o
r
m
a
l
i
s
e
d
D
e
s
o
r
p
t
i
o
n
R
a
t
e
(
K
-
1
)
Temperature (K)
Sigma = 5 kJ/mol
Sigma = 10 kJ/mol
Sigma = 15 kJ/mol
0.000
0.002
0.004
0.006
0.008
0.010
0.012
200 250 300 350 400 450 500
N
o
r
m
a
l
i
s
e
d
D
e
s
o
r
p
t
i
o
n
R
a
t
e
(
K
-
1
)
Temperature (K)
Beta = 0.1 K/s
Beta = 1 K/s
Beta = 10 K/s
22
following equation will hold: dT d dT d
A(g) A(ads)
u u = . Note, however, this equation is not generally
applicable to all scenarios. For example, in Reaction Set 2 discussed above and reproduced below, the rate
of production of
(g)
A does not equal dT d
A(ads)
u because
(ads)
A is also being converted to
(ads)
B .
(g) (ads) (ads) (g)
2B 2B A A
In future work, if readsorption is to be incorporated, then the concentration of gaseous species can be
simply worked out from the amount of gaseous species desorbing per second divided by the carrier gas
flow rate (assuming the amount of gas desorbing is small compared to the carrier gas flow rate, which is
normally the case).
TPD with Reaction:
In TPD with reaction, Equation 8 will have to be modified to account for multiple species present and for
multiple reactions occurring. As site points were indexed with the letter j and site types with the letter i
previously, reactions will be indexed with letter k and species with letter n, such that we will refer to
reaction k and to species n.
The rate of production of a species n due to reaction k occurring on site point j of site type i is given by
Equation 12 below. Recall that S is the stoichiometric matrix, such that
kn
S is the element of matrix S
corresponding to the stoichiometric coefficient of species n in reaction k. The species q in Equation 12
refers only to reactants involved in reaction k and
k
A refers to the pre-exponential factor of reaction k.
( )
(
=
[
|
u
u RT E A
S
dT
d
ij k
q
S
j i q kn
k j i n kq
exp
reactants
point , site , species
reaction , point , site , species
(12)
It is important to keep in mind that dT du is the rate of production. For example, if species n was a
product of reaction k then
kn
S would be positive, making dT du positive. However, if it was a reactant
then
kn
S would be negative, making dT du negative, indicating that species n is being consumed.
Equation 12 is only valid when the number of sites occupied by adsorbed products is less than or equal to
the number of sites occupied by adsorbed reactants. Otherwise, an extra term
sites empty
u raised to the power
of (number of adsorbed products number of adsorbed reactants) would need to be incorporated.
However, in order to avoid clutter, this will not be shown in the equations that follow, because it is not
applicable in the case studies analysed later on. Nevertheless, the model does accommodate for this term,
if appropriate.
Equation 12 only considered one reaction, but reactions occur simultaneously on each site point. Equation
13 below includes the contribution of all reactions.
23
( )
[
=
)
=
ions numOfReact
k
ij k
q
S
j i q kn
j i n
RT E A
S
dT
d
kq
1 reactants
point , site , species
point , site , species
exp
|
u
u
(13)
Finally, the rate of production from all site points of all site types must be added together to give the total
rate of production of species n, as shown in Equation 14 below.
= =
)
|
|
.
|
\
|
=
numOfSites
i
s numOfPoint
j
j i n
ij i
n
dT
d
dT
d
1 1
point , site , species species
u
c o
u
(14)
Therefore, the coverage of each species on each site point as a function of temperature will need to be
determined by numerical integration of Equation 13. This then allows calculation of the rate of production
of each species from each site point dT d
j i n point , site , species
u . Finally, this is plugged into Equation 14 to yield
the overall rate of production of each species dT d
n species
u as a function of temperature.
5. Model validation and discussion of results
This section will attempt to validate the model by applying it to several TPD experiments found in the
literature and to my partners experimental results for the isopropylamine/ZSM-5 system. Furthermore,
the current model will be compared to another independently produced model, in order to detect any
programming errors t. The following model validation is the result of my own work, with valuable help and
guidance from my supervisor Dr. Patrick Barrie. My partner did not contribute to validation of this model.
Experimental TPD data modelled include the following systems:
- Carbon monoxide desorption from a surface of Ni(111) (Miller, et al., 1987).
- Formic acid decomposition from a surface of Cu(110) (Ying & Madix, 1980).
- Isopropylamine reaction and desorption from zeolite ZSM-5.
Since the model does not consider readsorption, the first two case studies were chosen because the
experiments on metal crystal surfaces were conducted in ultra high vacuum, ensuring that readsorption
was negligible. The case study of CO on Ni(111) contains multiple overlapping peaks, but no reaction. The
case study of formic acid on Cu(110) has both reaction and desorption. The third case study was conducted
on a system in which readsorption is present; nonetheless, this scenario demonstrates the potential
usefulness and flexibility of the model, and gives an example of modelling TPD with reaction systems. If
readsorption is included into the model within future work, this case study may be revisited to obtain more
accurate results.
24
5.1 Comparison with specific model
At the beginning of this research project, a simplified model for TPD with reaction for isopropylamine on
zeolite ZSM-5 was developed. This model is specific to the isopropylamine/ZSM-5 system and assumes a
single activation energy for each reaction, rather than a distribution. The specific model contains the
reaction steps shown in Reaction Set 3 below.
The model assumes that propene is produced in the gaseous state rather than in an adsorbed state. This
assumption was made because of two reasons. Firstly, results of the experiment discussed in section 3.3
indicate that desorption of propene can be considered to be instantaneous for the purposes of modelling.
Secondly, isopropylamine bonds to Brnsted acid sites on ZSM-5 through the nitrogen atom, which means
when decomposition occurs, propene could be severed directly into a gaseous state (see Figure 15 below).
On the other hand, ammonia is modelled as being produced in an adsorbed state which may later desorb
because it is expected that nitrogen atom remain bonded to the site immediately after reaction. A program
to model this reaction scheme was written in Visual Basic and also in Java. The results of the general model
developed in this project could then be compared to these programs, in order to validate the model and
detect any programming errors.
H
3
C CH
3
Al
Si
+
NH
3
CH
O
-
Figure 15: Structure of adsorbed isopropylamine on a Brnsted
site.
(g) 3 (ads) 3
(ads) 3 (g) 2 3 (ads) 3 2 3
(g) 3 2 3 (ads) 3 2 3
NH NH 3.
NH CHCH CH )CH (NH CH CH 2.
)CH (NH CH CH )CH (NH CH CH 1.
Reaction Set 3: The set of reactions input into the current model
for comparison with the independent model.
The reaction set which was input into the current model is shown in Reaction Set 3 above. The value of
activation energy for each of the above reactions was set to be 100 kJ/mol. The pre-exponential factor was
set to be 10
13
s
-1
for each reaction and the initial coverage of isopropylamine was set to be unity. This
means that reactions 1 and 2 above will occur at the same rate; therefore, it is expected that the
desorption peaks for isopropylamine and propene will overlap completely. However, the peak for
ammonia will be delayed to higher temperatures because it is produced in the adsorbed state. The
coverage of ammonia is initially zero and reaction 2 needs time to produce enough ammonia such that the
rate of desorption of ammonia is appreciable. The simulated TPD curves for this situation are shown in
Figure 16 below. Note that both the general model and the specific program for this reaction predict
exactly the same results.
It is relevant to note here that the general model was also subjected to a large battery of other tests, in
which it performed as expected, but these tests will not be discussed in this report due to lack of space.
25
Figure 16: Both general and specific models predict the same results. Propene and isopropylamine curves are overlapping.
5.2 Case Study: CO on nickel
The desorption of carbon monoxide from a surface of Ni(111) was modelled using experimental data found
in the literature (Miller, et al., 1987). This system was chosen because readsorption is negligible and
because the results exhibit multiple overlapping peaks. Other quantitative methods struggle to extract
parameters from overlapping peaks. This example will demonstrate the strength of the model in dealing
with such complications. The process of fitting the model to experimental TPD data has not been
automated; therefore, a manual fitting procedure was conducted, explained in Figure 17 below. For
simpler cases with fewer variable parameters, such as that of formic acid on Cu(110), a more rigorous
sensitivity analysis could be conducted by also varying the pre-exponential factor (discussed later).
However, for the following case study of CO on Ni(111), the curves were fitted by eye because there were
too many parameters to conduct a manual optimisation (due to the presence of multiple peaks). Ideally
(and potentially in future work), an automated non-linear and multiple variable optimisation algorithm
could be used; however, for the purposes of demonstrating and validating the model, the approximate
procedure utilised is sufficiently accurate.
Define set of
reactions
Choose
number of
sites
Determine most
suitable pre-
exponential factor
from literature
Simulate TPD
0
0.002
0.004
0.006
0.008
0.01
0.012
230 280 330 380 430 480 530
R a t e o f D
e s o r p t i o n ( a r b i t r a r y u n i t s )
Temperature (K)
Is model
consistent with
experimental
results?
NO
YES
Adjust activation
energy
distribution and
site population
Guess activation
energy
distribution and
site population
Done
Figure 17: Process of fitting a model to experimental results.
Miller et al. (1987) published TPD experiments for the desorption of carbon monoxide adsorbed at
temperatures of 300 K and 87 K on Ni(111) , shown in Figures 18 and 19 below. For each adsorption
0
0.005
0.01
0.015
0.02
0.025
290 310 330 350 370 390 410
N
o
r
m
a
l
i
s
e
d
R
a
t
e
o
f
D
e
s
o
r
p
t
i
o
n
(
K
-
1
)
Temperature (K)
Isopropylamine (Specific)
Isopropylamine (general)
Ammonia (Specific)
Ammonia (General)
Propene (Specific)
Propene (General)
26
temperature, TPD was performed at different initial coverages. The saturation coverage for 300 K
adsorption was found to be 0.50 CO molecules per Ni atom (CO/Ni), while that for 87 K adsorption
corresponded to 0.57 CO/Ni. For CO adsorbed at 300 K, the main peak has a shoulder on the left. This can
be explained by the presence of at least two binding states of similar binding energy for the CO molecule
on Ni(111). A new low temperature peak appears for CO adsorbed at 87 K, which corresponds to a new
lower binding energy state of the CO molecule. Furthermore, as the initial coverage is increased, the peak
temperature can be seen to decrease. Since desorption of CO on nickel is expected to be first-order
(discussed in the next paragraph), this can only be explained by the decrease of activation energy with
increasing coverage i.e. adsorbed CO molecules interact unfavourably with each other. Therefore, the
activation energy is expected to be a function of coverage, which was confirmed by Christmann et al.
(1974). Also, the high energy binding state is occupied before the low energy binding state at low
coverages, indicating that CO is mobile on the surface of Ni(111).
Figure 18: TPD of CO adsorbed on Ni(111) at 300 K performed at
a heating rate of 6 K/s (Miller, et al., 1987).
Figure 19: TPD of CO adsorbed on Ni(111) at 87 K performed at a
heating rate of 6 K/s (Miller, et al., 1987).
Simple desorption of CO is expected as shown in Reaction Set 4 below which was used to model the
experimental data. However, CO is known to dissociate and disproportionate on some transition metals
(Steinruck, et al., 1986); therefore, it is imperative to determine whether dissociation occurs on Ni(111) in
the temperature range sampled as this will affect the reaction set used in the model. Tomanek &
Bennemann (1983) determined that the activation energy for dissociation of CO on Ni(111) is 209 kJ/mol,
which means this reaction will only be significant at temperatures above 600 K (determined by simulating a
first-order dissociation reaction with E
act
= 209 kJ/mol). Given that nearly all CO desorbs at a temperature
significantly lower than 600 K on Ni(111), the dissociation surface reaction may be safely neglected. On a
further note, Nakamura et al. (1989) support this by saying that dissociation does not occur at the
desorption temperatures for CO on Ni(111). Another possible factor is the disproportionation reaction
27
2CO
(ads)
C
(ads)
+ CO
2
, where two CO molecules combine to produce an adsorbed surface carbon species
and CO
2
. However, when CO is adsorbed at temperatures lower than 300 K, molecular CO desorbs when
heated to 500 K without any trace of adsorbed carbon or oxygen, indicating that neither dissociation nor
disproportionation occurs within the temperature range of the experiment (Steinruck, et al., 1986).
(g) (ads)
CO CO
Reaction Set 4: Simple desorption of CO.
The TPD data for adsorption at 300 K and saturation coverage of 0.50 CO/Ni was modelled using two sites
(site 1 = low temperature shoulder, site 2 = high temperature main peak), each with a distribution of
activation energies.
The pre-exponential factor used for desorption of CO was taken to be A = 10
13
s
-1
because this is the value
expected for unimolecular desorption it corresponds to the frequency of a bond vibration. In any case, an
order of magnitude change in A affects the activation energy by only 5 % (Barrie, 2012). The parameters
that were varied to fit experimental data are shown in Table 4 below along with their optimised values
(note that a fit was not possible unless a distribution of activation energy was used for each site):
Table 4: Parameters used to fit the model to experimental TPD data for absorption at 300 K.
E
mean
(kJ/mol) (kJ/mol)
Site 1 99.3 8.9 0.38
Site 2 115.7 5.4 0.62
Note that
1
+
2
must always equal 1. Figure 20 shows how the model compares to experimental TPD data
observed by Miller et al. (1987). The fit was good with an error of about 0.9 % (see Equation 16 below),
especially given that there were five independent parameters to vary and that it was done manually.
( ) =
2
Experiment Model Error Square of Sum
(15)
( )
( )
100
2
2
=
Experiment
Experiment Model
Percentage Error
(16)
Figure 20: Comparison of model to experimental TPD data for CO
adsorption at 300 K on Ni(111).
Figure 21: Optimisation of pre-exponential factor on TPD curve
of initial coverage equal to 0.032 CO/Ni for 300 K adsorption of
CO on Ni(111). The sum of square error is given in Equation 15.
The line drawn is to guide the eye.
28
The peak temperature of the high binding energy peak of CO (i.e. site 2) was determined to be 444 K from
the experimental data in Figure 20. Redheads method (described in Section 3) was applied using this peak
temperature and a pre-exponential factor of 10
13
s
-1
, and the resulting activation energy was determined
to be 113.7 kJ/mol. This compares very well to our value of E
mean, 2
= 115.7 kJ/mol. Furthermore,
Christmann et al. (1974) determined an activation energy for desorption of 111 kJ/mol at medium
coverages. None of the other quantitative methods can be easily applied to compare values for the
activation energy of the low binding energy peak (i.e. site 1) due to the two peaks overlapping.
During the TPD experiment, coverage of CO gradually decreases; therefore, the activation energy is
expected to change accordingly. However, in the limit of low coverage (see Figure 18 above) a single peak
is observed for the high energy binding state of CO (site 2). Miller et al. (1987) used the peak width analysis
method (described in Section 3) to determine an activation energy of 128 kJ/mol and a pre-exponential
factor of 1.4 x 10
14
s
-1
, for a single peak at an initial coverage of 0.04 CO/Ni. As expected, the activation
energy is higher for lower coverages.
The standard deviation of activation energy gives, in this case, a quantitative indication of how much the
activation energy varies with coverage. In fact the activation energy determined by Miller et al. (1987) for
the high binding energy state at very low coverage is within E
mean, 2
+ 2.3
2
. This agrees well with our
predictions because values of activation energy at 2.3
2
are at the outskirts of the distribution, in line with
the fact that Miller et al. (1987) calculated the activation energy in the limit of very low coverage.
Furthermore, Christmann et al. (1974) reported values of the isosteric heat of adsorption E
st
of CO on
Ni(111), which is related to the activation energy of desorption by E
act
= E
st
- 0.5RT, for the non-activated
adsorption of CO (Miller, et al., 1987). The values calculated for the activation energy of desorption from
the results of Christmann et al. (1974) over a wide range of coverages are approximately 81.9
126.4 kJ/mol, which corresponds to E
mean, 1
- 2
1
and E
mean, 2
+ 2
2
, this also supports our findings since
values within E
mean
2 contain about 95 % of the distribution.
In order to better compare the model results to the peak width analysis method used by Miller et al.
(1987), the model was fitted to the low coverage peak of 0.032 CO/Ni in Figure 18 using a single site
without a distribution of activation energy. In this case only two parameters needed variation (E
act
and A);
therefore, a more rigorous optimisation procedure was followed, shown in Figure 21 above. For every
value of A plotted, the value of E
act
was optimised by minimising the sum of the square error between the
model and experimental data. As a result, the best fit parameters deduced were approximately A = 10
13
s
-1
and E
act
= 119.7 kJ/mol. This further supports our use of 10
13
s
-1
as the pre-exponential factor. The different
values between our model and the peak width method used by Miller et al. (1987) may be due to the fact
29
that there was a fitting error of approximately 9.4 %. This error can explain the discrepancy in activation
energy reported. It is believed that a good fit was not obtained in this case because a single activation
energy was used, rather than a distribution, given that Christmann et al. (1974) reported a sudden, large
variation in activation energy for small coverages. Furthermore, two types of site (two-fold and three-fold
bridged sites) have been suggested to occur at very low coverages of CO on Ni(111) (Surnev, et al., 1988)
which may also explain the discrepancy.
The experimental TPD curve at saturation coverage (0.57 CO/Ni) for adsorption at 87 K (see Figure 19) was
also modelled, but this time using three sites to account for the new lower energy binding state that
appeared (see Figure 22 for comparison of the model with experimental data). The fit was good with an
error of about 0.7 %. The parameters determined are summarised in Table 5 below. The sites are labelled
in Figure 23, which shows the rate of desorption from each site separately, as predicted by the model.
Figure 22: Comparison of model with experimental results for CO
adsorbed at 87 K.
Figure 23: Rate of desorption of CO, as predicted by the model,
for each site. The total desorption rate is also shown.
Table 5: Summary of parameters used to model CO adsorbed at 87 K with an initial coverage of 0.57 CO/Ni.
E
mean
(kJ/mol) (kJ/mol)
Site 1 102 9.9 0.402
Site 2 115 4.8 0.493
Site 3 75.4 5.9 0.105
The activation energies and standard deviations for sites 1 and 2 were approximately the same as for CO
adsorbed at 87 K and 300 K. Miller et al. (1987) also discovered similar activation energies at the limit of
low coverage for adsorption at 300 K and 87 K. The peak temperature of site 3 is difficult is discern;
however, an approximate value is 293 K. Redheads method was used using this peak temperature (and
A = 10
13
s
-1
) to determine an activation energy of 74.1 kJ/mol for site 3. This compares very well with our
value of 75.4 kJ/mol.
0.000
0.002
0.004
0.006
0.008
0.010
0.012
200 300 400 500
N
o
r
m
a
l
i
s
e
d
D
e
s
o
r
p
t
i
o
n
R
a
t
e
Temperature (K)
Model
Experiment
30
A key quantitative result of the modelling process is the population fraction of sites present on the catalyst,
a figure that is usually difficult to obtain from overlapping peaks. It is interesting to note that the
population fraction of site 2 dropped significantly from
2
= 0.62 at an adsorption temperature of 300 K to
2
= 0.49 at an adsorption temperature of 87 K. The population fraction of site 1 increased slightly from
1
= 0.38 to
1
= 0.40. In addition, the saturation coverage increased from 0.50 CO/Ni to 0.57 CO/Ni. This
means that some CO molecules in the high energy binding state of site 2 are moving into the lower binding
energy state of sites 1 and 3. These results can be explained by the fact that Surnev et al. (1988) and Tang
et al. (1986) reported the presence of two-fold bridged and terminal CO sites on Ni(111), which are
illustrated in Figures 24(a) and 24(b) below. In particular, it was reported the relative populations of two-
fold bridged sites to terminal sites is both temperature and coverage dependent. Also, the two-fold
bridged site has a stronger binding energy than the terminal site (Tang et al., 1986). This leads us to believe
that site 1 is mainly due to desorption from terminally bound CO molecules, while site 2 is that due to two-
fold bridged molecules. Site 3 may be attributed to neighbouring CO species at high loadings, because
there will be unfavourable interactions between neighbouring CO species due to the partial negative
charge of CO on Ni(111) (Somorjai, 1994), as shown in Figure 24(c). In support, King (1975) notes that
repulsion between adsorbed species can cause multiple TPD peaks from a crystal metal surface. The
population fraction determined from the model may also be used to validate quantitatively proposed
binding models of CO on Ni(111); however, due to a lack of time and space, this was not performed.
Figure 24: (a) Terminal site (b) Two-fold bridged site (c) Unfavourable interactions between neighbouring CO molecules at high surface
coverages.
The model developed has proven to be useful and accurate in determining the range of activation energies
for CO desorption from multiple overlapping sites, which is normally difficult to perform using other
quantitative methods. The population fraction of sites was also determined, which allowed insights to be
made on the binding structure of CO on Ni(111).
5.3 Case Study: formic acid on copper
TPD with reaction of formic acid adsorbed onto a crystal surface of Cu(110) was modelled using
experimental results published in the literature. The reaction mechanism for formic acid decomposition on
Cu(110) has been studied by Ying & Madix (1980) and is shown below in Figure 26. Formic acid adsorbs
dissociatively to produce an adsorbed hydrogen atom
(ads)
H and formate species
(ads)
HCOO . As the
temperature is increased during TPD, the adsorbed hydrogen atoms present recombine to desorb as
31
molecular hydrogen. At yet higher temperatures, the formate species decomposes to produce gaseous
carbon dioxide and an adsorbed hydrogen atom, which desorbs rapidly because the temperature at which
formate decomposition occurs is much higher than that for hydrogen desorption. The experimental TPD
results used are shown in Figure 25 below.
Figure 25: TPD experiment of formic acid on Cu(110) using a
heating rate of = 3.4 K/s (Ying & Madix, 1980).
(ads) (g) 2 (ads)
2(g) (ads)
(ads) (ads) (g)
H CO HCOO 3.
H 2H 2.
H HCOO HCOOH 1.
+
+
Figure 26: Reaction mechanism for Formic Acid on Cu(110).
As can be seen, adsorbed hydrogen atoms that formed upon adsorption of formic acid desorb at low
temperature; furthermore, the hydrogen formed due to decomposition of the formate species is
coincident with the evolution of carbon dioxide [carbon dioxide cannot adsorb on Cu(110) because it does
not have a binding state (Ying & Madix, 1980)]. The low temperature hydrogen peak was modelled
separately to the other peaks, and two important issues were discovered:
- Due to the stoichiometry of the process, the low temperature hydrogen peak should have an area
equal to that of the high temperature hydrogen peak. However, it was realised that the area of the
low temperature hydrogen peak is in fact 51% of the high temperature peak. Ying & Madix (1980)
also realised this discrepancy (but they quoted it as being 70%) and commented that during
adsorption, formate species may compete with adsorbed hydrogen atoms for available sites, which
may cause some hydrogen to desorb upon adsorption of formic acid, explaining this apparent
inconsistency. A more agreeable observation was that the area of the high temperature hydrogen
peak equals almost exactly half that of the CO
2
peak, as expected by stoichiometry.
- The shape of the hydrogen peak is somewhat triangular and does not correspond to that expected
for second-order desorption kinetics. The presence of formate species on the surface is known to
cause a reduction in the desorption temperature for hydrogen (Wachs & Madix, 1979) indicating
that formate species do affect hydrogen desorption. This interaction may account for the irregular
shape observed. The irregular shape makes fitting a curve difficult, as discussed below.
The reaction set used for modelling the low-temperature hydrogen peak consists of one reaction
2H
(ads)
H
2(g)
. The model used consists of one type of site with a single activation energy, rather than a
32
distribution, because it was found that if a distribution was used the pre-exponential factor obtained is
unreasonably high. Furthermore, a uniform surface of Cu(110) contains only one type of site and this
supports the use of a single activation energy. A manual optimisation fitting by altering both the pre-
exponential factor and the activation energy was not useful in this case because the irregular shape of the
low temperature hydrogen peak resulted in similar fittings for all cases. Consequently, a pre-exponential
factor of 4 x 10
8
s
-1
was chosen because Wilmer et al. (2003) reported this value for desorption of
hydrogen from a supported copper catalyst. The activation energy was optimised with respect to the
chosen pre-exponential factor and was found to be 49.8 kJ/mol; the optimisation curve is shown in Figure
27 below. The model and experimental TPD curves are given in Figure 28 below and as can be seen, an
exact fit was not possible (fitting error of about 16 %) due to the triangular shape of the experimental
curve and the human error involved in reading experimental data from published graphs.
Figure 27: Optimisation with respect to activation energy of
desorption for the low temperature hydrogen peak, assuming a
pre-exponential factor of 4 x 10
8
s
-1
. The line drawn is to guide
the eye.
Figure 28: Model and experimental TPD curves for the low
temperature hydrogen peak from formic acid on Cu(110).
On a supported copper catalyst, Wilmer et al. (2003) reported the activation energy for desorption of
hydrogen to be 58 kJ/mol. The reduced activation energy we determined in the presence of formate
species is supported by the fact that Ying & Madix (1980) reported a significantly lower peak temperature
(about 50 K lower) for hydrogen in the presence of formate species on Cu(110). Furthermore, Wachs &
Madix (1979) reported an activation energy of 50.2 4.2 kJ/mol for D
2
from a Cu(110) surface, this
compares very well to our value of 49.8 kJ/mol. Finally, Redheads method for quantitative analysis on the
low temperature hydrogen curve (using T
p
= 274 K and
p
u equal to half the initial coverage) results in a
calculated activation energy of 48.2 kJ/mol, which is also similar to our value.
The decomposition of formate species was modelled using Reaction Set 5 shown below. The kinetic
parameters resolved from the low temperature hydrogen peak were used for reaction 2 in this model. In
this case, the CO
2
curve could not be modelled using a distribution of activation energies because the
33
simulated curve would not fit experimental results unless a very small standard deviation was used i.e. a
single activation energy was needed. This is supported by the fact that a uniform Cu(110) surface contains
only one type of site. Not having a distribution of activation energies facilitates the manual simultaneous
optimisation of both the pre-exponential factor and the activation energy for formate decomposition. This
was performed by first selecting a pre-exponential factor and then conducting an optimisation for
activation energy, as done with hydrogen above. Numerous values for the pre-exponential factor were
tested, producing Figure 29 below. This resulted in an optimum pairing of A = 7.5 x 10
15
s
-1
and
E
act
= 149.8 kJ/mol. The model and experimental results for both the CO
2
and high temperature H
2
peaks
are shown in Figure 30 below. Note that the CO
2
peak shows a good fitting, with an error of 2.4 %.
(g) 2 (ads)
(ads) (g) 2 (ads)
H 2H 2.
H CO HCOO 1.
+
Reaction Set 5: Decomposition of adsorbed formate.
Figure 29: Optimisation of A for formate reaction. E
act
was
optimised for each plotted point above. The line drawn is to
guide the eye.
Figure 30: Model and experimental TPD curves for the CO
2
and
high temperature H
2
peaks from formic acid on Cu(110).
Ying & Madix (1980) normalised the high-temperature hydrogen and carbon dioxide peaks and showed
that they overlap precisely, as expected because formic acid decomposition is rate limiting. However, when
this was performed using data read from the published graphs, the hydrogen curve overshot the CO
2
curve
slightly. This proves that error in extracting data points from graphs over 30 years old is unavoidable. As a
result, the experimental hydrogen curve overshoots the model, as can be seen in Figure 30 above.
Ying & Madix (1980) conducted quantitative analysis on formic acid decomposition using three methods:
variation of heating rate, variation of initial coverage and fitting a model to experimental data. The results
reported were A = 9.4 x 10
13
s
-1
and E
act
= 133.3 kJ/mol. In order to illustrate why our parameters differed
from those of Ying & Madix (1980), Figure 31 shows a plot of ln(rate constant) vs. 1/T for our values of A
and E
act
and for those of Ying & Madix (1980) over a temperature range of 425-500 K. As can be seen, both
sets of parameters yield very similar values for the rate constant. This is because it is possible to have
34
different pairs of A and E
act
that predict similar values for the rate constant. When the same
pre-exponential factor of A = 9.4 x 10
13
s
-1
used by Ying & Madix (1980) was input into our model and fitted
to the experimental CO
2
curve in Figure 25, the resulting optimised activation energy was found to be
E
act
= 132.8 kJ/mol. This compares very well to the value of 133.3 kJ/mol reported by Ying & Madix (1980).
Figure 31: Comparison of the rate constant calculated using the parameters determined from our model (A = 7.5 x 10
15
s
-1
and
E
act
= 149.8 kJ/mol) and those determined using the results of Ying & Madix (1980) (E
act
= 133.3 kJ/mol, A = 9.4 x 10
13
s
-1
).
5.4 Case Study: Isopropylamine on ZSM-5
My partners experimental TPD data for isopropylamine on ZSM-5 (with full initial coverage) is shown in
Figure 32 below (after correcting for baseline drift). The m/e ratios of 44, 41 and 17 correspond to the
strongest peaks in the spectra of isopropylamine, propene and ammonia, respectively. However, it is
important to note that isopropylamine produces a m/e = 41 fragment in the mass spectrometer as well.
0
2E-10
4E-10
6E-10
8E-10
1E-09
1.2E-09
300 400 500 600 700 800 900 1000
M
a
s
s
S
p
e
c
t
r
o
m
e
t
e
r
S
i
g
n
a
l
(
a
u
)
Temperature (K)
m/e = 44
m/e = 41
m/e = 17
Figure 32: TPD of isopropylamine on ZSM-5 with full initial
coverage and a heating rate of 0.17 K/s (Miao, 2012).
0.000
0.001
0.002
0.003
0.004
0.005
0.006
0.007
0.008
0.009
300 350 400 450 500 550 600
N
o
r
m
a
l
i
s
e
d
D
e
s
o
r
p
t
i
o
n
R
a
t
e
(
K
-
1
)
Temperature (K)
Model
m/e = 41
Figure 33: Comparison of fitted model to experimental results for
isopropylamine desorption.
Isopropylamine desorbs between 320 K and 585 K; however due to noise in the signal, it is difficult to
model the m/e = 44 curve. It is apparent from Figure 32 and from others (Parrillo, et al., 1990) that
-7
-6
-5
-4
-3
-2
-1
0
1
0.0019 0.002 0.0021 0.0022 0.0023 0.0024
l
n
(
r
a
t
e
c
o
n
s
t
a
n
t
)
1/T (K
-1
)
Model
Ying & Madix
35
propene desorbs only slightly between 550 K and 585 K. Therefore, the m/e = 41 fragment of
isopropylamine will be used (after normalisation and correction for slight propene desorption) to extract
the kinetic parameters for desorption of isopropylamine. A shoulder on the right of the m/e = 41 curve
indicates that there are at least two sites from which isopropylamine desorbs. It is expected that ZSM-5
contains some weak acid sites with a distribution of activation energy because the pore structure contains
two types of channels one straight and one zig-zag (Structure Commission of the International Zeolite
Association, 2008). Moreover, sites located at intersections between channels might have yet another
binding energy. As a result, desorption of isopropylamine is modelled using simple desorption from two
sites, each with a distribution of activation energy. The pre-exponential factor for desorption was taken to
be A = 10
13
s
-1
, as expected for unimolecular desorption (Barrie, 2012). The model was fitted to the
normalised and corrected m/e = 41 curve with an error of 0.24 %, and is shown in Figure 33 above. The
kinetic parameters extracted for both sites are shown in Table 6 below. The only other quantitative
method that can be applied (with caution) to overlapping peaks is Redheads method. However, in this
case only the peak temperature of site 1 can be distinguished to be about 410 K. Redheads method (with
A = 10
13
s
-1
, | = 0.17 K/s) gives an activation energy of 116.6 kJ/mol, which compares well with our value
of 118.4 kJ/mol. Our simulations show that both sites have similar standard deviations of 8.8 kJ/mol and
8.7 kJ/mol, indicating that the distribution of activation energies for each site varies over a range of about
2 = 17.5 kJ/mol.
Table 6: Summary of kinetic parameters extracted for
isopropylamine desorption.
E
mean
(kJ/mol) (kJ/mol)
Site 1 118.4 8.8 0.57
Site 2 139.1 8.7 0.43
(g) 3 (ads) 3
(ads) 3 (g) 2 3 (ads) 3 2 3
NH NH 2.
NH CHCH CH )CH CH(NH CH 1.
+
Reaction Set 6
Figure 32 shows that reaction to produce propene and ammonia takes place between 550 K and 650 K. The
propene peak has a shape consistent with a first-order reaction (as expected); however, the ammonia peak
is shifted to higher temperatures and has a long tail. The heat of adsorption for ammonia on ZSM-5 was
reported to be 150 kJ/mol by Parrillo & Gorte (1992), which is expected to equal the activation energy of
desorption because the adsorption process is not activated. This means that a peak temperature of 550 K
is expected for simple desorption of ammonia from ZSM-5; however, Figure 32 shows a peak of about
650 K for ammonia. Given the fact that propene desorbs significantly sooner than ammonia, we know that
the reaction is not limiting the desorption of ammonia. This indicates that the emergence of ammonia is
controlled by readsorption. The propene peak will thus be modelled to extract kinetic parameters for the
reaction of isopropylamine. The strong Brnsted sites are expected to have a full coverage of
isopropylamine until reaction occurs (see Section 2.4). We can therefore perform simulations using one
36
site with full initial coverage of isopropylamine. Reaction Set 6 above was input into the model for reasons
given in Section 5.1. Since the model does not consider readsorption, the peak shape of ammonia cannot
be simulated by assuming first-order desorption of ammonia. As a result, the ammonia curve cannot be
fitted to the model and the parameters for ammonia desorption cannot be determined. The model and
experimental propene curves are shown in Figure 34 below, the fitting error was about 8 %. It was found
that a good fit could not be attained with the propene peak (using a pre-exponential factor of A = 10
13
s
-1
)
unless a single activation energy was used i.e. no distribution. This is supported by the fact that
experimental evidence shows that these Brnsted sites on ZSM-5 are of equal strength i.e. will give equal
activation energies (Parrillo & Gorte, 1992; Kresnawahjuesa, et al., 2002). This enabled a more rigorous
optimisation procedure for A to be performed as shown in Figure 35 below. The activation energy for the
isopropylamine reaction was determined to be 187.9 kJ/mol with a pre-exponential factor of
A = 5 x 10
13
s
-1
. Redheads method gives an activation energy of 187.3 kJ/mol, which compares very well to
our value. Note that the activation energy for reaction is lower than that for isopropylamine desorption
from Brnsted sites (240 kJ/mol, see Section 2.4), which is as expected because decomposition is observed
to occur before desorption.
0.000
0.005
0.010
0.015
0.020
0.025
540 560 580 600 620 640 660
N
o
r
m
a
l
i
s
e
d
D
e
s
o
r
p
t
i
o
n
R
a
t
e
(
K
-
1
)
Temperature (K)
Model
Experiment
Figure 34: Plot of model and experiment for the propene curve.
Figure 35: Optimisation of pre-exponential factor for reaction of
isopropylamine. The line drawn is to guide the eye.
0.0E+00
5.0E-06
1.0E-05
1.5E-05
2.0E-05
2.5E-05
3.0E-05
3.5E-05
4.0E-05
1.E+12 1.E+13 1.E+14 1.E+15
S
u
m
o
f
S
q
u
a
r
e
E
r
r
o
r
Pre-exponential Factor (s
-1
)
37
6. Conclusion
A general model for temperature programmed desorption (TPD) with reaction has been developed and
successfully validated. The model can cope with multiple overlapping sites and can attribute a distribution
of activation energies to each site. The flexibility of the model in allowing the specification of a set of
reactions to describe any system proved to be very useful. Quantitative information such as distribution of
activation energies for each site, pre-exponential factors, and relative site populations, may be determined
by fitting the model to experimental TPD data. Three case studies were undertaken, from which
quantitative parameters were extracted that agreed well with values reported in the literature. In order to
understand better the underlying dynamics of one of the case studies (isopropylamine/ZSM-5), some
experiments were performed to determine the temperature range of propene desorption from ZSM-5 and
the extent of thermal decomposition of propene. The ability of the model to extract parameters from TPD
with reaction experiments and the strength of the model in dealing with multiple overlapping peaks was
demonstrated, a feat rarely accomplished using other methods.
7. Further work
Future work that may be performed in continuation of this research include:
- Incorporation of readsorption effects into the model.
- TPD experiments of ammonia on ZSM-5 in order to model more accurately the
isopropylamine/ZSM-5 system.
- Case studies involving more complicated reaction systems such as CO on a supported-ruthenium
catalyst, on which I have already done significant work, but did not have space to write about in this
report.
8. Nomenclature
English
A Pre-exponential factor in rate constant 1/s
E Activation energy J/mol
isAdsorbed Matrix storing state of species (adsorbed or gaseous) -
k Rate constant 1/s
m/e Mass to charge ratio -
n Order of desorption/reaction -
numOfMolecules Number of species in model -
numOfPoints Number of site points considered for the distribution on each site type -
numOfReactions Number of reactions in model -
numOfSites Number of site types in model -
R Universal gas constant J/mol K
T Temperature K
r Rate of Desorption 1/s
S Stoichiometric matrix -
38
t Time s
W Width of desorption peak K
Greek
Fractional propulation of site types matrix -
Heating rate during TPD K/s
Fractional weighting matrix -
Coverage -
Standard deviation of distribution J/mol
Subscripts
1/2 Half peak rate of desorption conditions
3/4 Three-quarters peak rate of desorption conditions
act Activation
f Final
i Corresponding to site type i
j Corresponding to site point j
k Corresponding to reaction k
mean Mean value of distribution
n Corresponding to species n
o Initial
p Peak rate of desorption conditions
st Isoteric heat of adsorption
9. Works cited
(NIST), National Institute of Standards and Technology, 2011. NIST Chemistry WebBook. [Online]
Available at: http://webbook.nist.gov/chemistry/
[Accessed 25 4 2012].
Anon., 2008. LookChem. [Online]
Available at: http://www.lookchem.com/Isopropylamine/
[Accessed 1 5 2012].
Barrie, P., 2012. CET IIB Catalysis Lecture Notes. s.l.:s.n.
Brown, A. & Vickerman, J. C., 1981. Surface coverage measurements for CO adsorption on Ru(001); a combined
SIMS/TPD study. Vacuum, 31(10-12), pp. 429-433.
Chan, C., Aris, R. & Weinberg, W., 1978. An Analysis of Thermal Desorption Mass Spectra. Application of Surface
Science, Volume 1, pp. 360-376.
Christmann, K., Schober, O. & Ertl, G., 1974. Adsorption of CO on a Ni(111) surface. The Journal of Chemical Physics,
Volume 60, pp. 4719-4724.
Criado, J. M., Malet, P., Munuera, G. & Rives-Arnau, V., 1982. Remarks on "Temperature-Programmed Desorption
from Porous Catalysts: Shape Index Analysis". Journal of Catalysis, Volume 75, pp. 428-432.
Cvetanovic, R. J. & Amenomiya, Y., 1972. A Temperature Programmed Desorption Technique for Investigation of
Practical Catalysts. Catalysis Reviews: Science and Engineering, 6(1), pp. 21-48.
39
Falconer, J. & Madix, R., 1977. Desorption Rate Isotherms in Flash Desorption Analysis. Journal of Catalysis, Volume
48, pp. 262-268.
Falconer, J. & Schwarz, J., 1983. Temperature-Prgrammed Desorption and Reaction: Application to Supported
Catalysts. Catalysis Reviews, 25(2), pp. 141-227.
Gorte, R., 1982. Design Paramters for Temperature Programmed Desorption from Porous Catalysts. Journal of
Catalysis, Volume 75, pp. 164-174.
Ibok, E. & Ollis, D., 1980. Temperature-Programmed Desorption from porous Catalysts: Shape Index Analysis. Journal
of Catalysis, Volume 66, pp. 391-400.
Jong, A. d. & Niemantsverdriet, J., 1990. Thermal Desorption Analysis: Comparitive Test of Ten Commonly Applied
Procedures. Surface Science, Volume 233, pp. 355-365.
King, D., 1975. Thermal Desorption from Metal Surfaces: A Review. Surface Sciences, Volume 47, pp. 384-402.
Kresnawahjuesa, O., Kuhl, G. H., Gorte, R. J. & Quierini, C. A., 2002. An Examination of Bronsted Acid Sites in H-
[Fe]ZSM-5 for Olefin Oligomerization and Adsorption. Journal of Catalysis, Volume 210, pp. 106-115.
Laidler, K. J. & Wojciechowski, B. W., 1960. Kinetics of the Thermal Decomposition of Propylene, and of Propylene-
Inhibited Hydrocarbon Decompositions. Proceedings of the Royal Society of London. Series A, Mathematical and
Physical Sciences, 259(1297), pp. 257-266.
Lee, P. I. & Schwarz, J. A., 1982. Adsorption-Desorption Kinetics of H2 from Supported Nickel Catalysts. Journal of
Catalysis, Volume 73, pp. 272-287.
Low, G. & Bell, A., 1979. Studies of CO Desorption and Reaction with H2 on Alumina-Supported Ru. Journal of
Catalysis, Volume 57, pp. 397-405.
McCarty, J. & Wise, H., 1979. Dissociative Chemisorption of CO on Ruthenium. Chemical Physics Letters, 61(2), pp.
323-326.
Miao, Q., 2012. Temperature Programmed Desorption with Reaction (IIB Research Report), Cambridge: s.n.
Miller, J. B. et al., 1987. Extraction of kinetic parameters in temperature programmed desorption: A comparison of
methods. The Journal of Chemical Physics, 87(11), pp. 6725-6732.
Nakamura, J. et al., 1989. Fromation of a Hybrid Surface of Carbide and Graphite Layers on Ni(100) but no Hybrid
Surface on Ni(111). Surface Science, Volume 222, pp. L809-L817.
Niwa, M. & Katada, N., 1997. Measurements of acidic property of zeolites by temperature programmed desorption
of ammonia. Catalysis Surveys from Japan, Volume 1, pp. 215-226.
Parrillo, D., Adamo, A., Kokotailo, G. & Gorte, R., 1990. Amine adsorption in H-ZSM-5. Applied Catalysis, Volume 67,
pp. 107-118.
Parrillo, D. & Gorte, R., 1992. Charaterization of stoichiometric adsorption complexes in H-ZSM-5 uning
microcalorimetry. Catalysis Letters, Volume 16, pp. 17-25.
Parrillo, D. J., Dolenec, D., Gorte, R. J. & McCabe, R. W., 1993. Adsorption Studies on Cu-ZSM-5: Characterization of
the Unique Properties of Ion-Exchanged Cu. Journal of Catalysis, Volume 142, pp. 708-718.
Redhead, P., 1962. Thermal desorption of gases. Vacuum, 12(4), pp. 203-211.
40
Somorjai, G., 1994. Introduction to surface chemistry and catalysis. New York: Wiley-Interscience.
Steinruck, H., D'Evelyn, M. & Madix, R., 1986. The Role of Defects in the Dissociative Adsorption of CO on Ni(100).
Surface Science Letters, Volume 172, pp. L561-L567.
Structure Commission of the International Zeolite Association, 2008. Database of Zeolite Structures. [Online]
Available at: http://izasc.ethz.ch/fmi/xsl/IZA-SC/ftc_fw.xsl?-db=Atlas_main&-lay=fw&-max=25&STC=MFI&-find
[Accessed 11 5 2012].
Surnev, L., Xu, Z. & Yates, J., 1988. IRAS Study of the Adsorption of CO on Ni(111): Interrelation Between Various
Bonding Modes of Chemisorbed CO. Surface Science, Volume 201, pp. 1-13.
Tang, S. L. et al., 1986. Bridge/atop site coversion of CO on Ni(111): Determination of the binding energy difference.
The Journal of Chemical Physics, Volume 1883, p. 1876.
Tomanek, D. & Bennemann, K., 1983. LCAO-Type Calculations of the Effect of K and Cl Atoms on the Dissocation of
CO at the Ni(111) Surface.. Surface Science, Volume 127, pp. L111-L117.
Wachs, I. & Madix, R., 1979. The Oxidation of H2CO on a Copper(110) Surface. Surface Science, Volume 84, pp. 375-
386.
Wilmer, H., Genger, T. & Hinrichsen, O., 2003. The interaction of hydrogen with alumina-supported copper catalysts:
a temperature-programmed adsorption/temperature-programmed desorption/isotopic exchange reaction study.
Journal of Catalysis, Volume 215, pp. 188-198.
Ying, D. & Madix, R., 1980. Thermal Desorption Study of Formic Acid Decomposition on a Clean Cu(110) Surface.
Journal of Catalysis, Volume 61, pp. 48-56.
41
10. Safety appendix
All possible precautions were taken to minimise the risks involved while performing experiments. The risks
involved and precautions undertaken are listed below:
- High pressure gas cylinders
Only qualified personnel were allowed to handle gas cylinders. Regulators and pressure gauges
were used at all times. The cylinders were kept secured and stable to prevent them from
accidentally falling. Cylinder valves were always closed when not in use.
- Flammable (propene and isopropylamine)
The flow rate of propene was kept to a minimum (about 2 ml/min). Propene was either combusted
in a safe manner or discharged externally to the environment. Possible sources of ignition in the
laboratory were avoided.
- Asphyxiation (helium gas)
A low flow rate of helium was maintained (40 ml/min); therefore, this did not constitute a
significant risk of asphyxiation. Nevertheless, we were made very aware of the risk of an accidental
gas leak and a gas sensor alarm was always in use in the laboratory, as a precautionary measure.
- Toxic/irritant substances (isopropylamine and organic solvents)
Isopropylamine is a volatile liquid at room temperature; therefore, it was necessary to handle the
substance in a fume cupboard. This minimised the risk of lung irritation caused by isopropylamine
vapours. In addition, nitrile gloves were used at all times when handling such toxic or irritant
substances to avoid skin contact.
The risks involved in developing the model include back ache, repetitive strain injury, and computer eye
strain. The steps taken to avoid such problems involved taking frequent breaks with occasional stretching
in order to relive muscle tension.
Вам также может понравиться
- CHM 510 Exp 1 GCДокумент8 страницCHM 510 Exp 1 GCNurul HaziqahОценок пока нет
- Liquid-Liquid Extraction Technique GuideДокумент36 страницLiquid-Liquid Extraction Technique GuideamirnimoОценок пока нет
- Chm557 Exp2Документ4 страницыChm557 Exp2Rap DutaОценок пока нет
- Lab Report 3.0Документ7 страницLab Report 3.0Husna Insyirah Bt SamadОценок пока нет
- Hardness of WaterДокумент6 страницHardness of WaterJamesShiq0% (1)
- Experiment 1 Optimization of Flow Rate and Column Temperature (Method Development)Документ7 страницExperiment 1 Optimization of Flow Rate and Column Temperature (Method Development)NUR IZZATI OTHMAN BASRIОценок пока нет
- COD Test Determines Organic PollutantsДокумент4 страницыCOD Test Determines Organic Pollutantskh!mОценок пока нет
- Assignment Tokoh Organometallic GilmanДокумент7 страницAssignment Tokoh Organometallic GilmanniniОценок пока нет
- Determination of The Percentage of Ligands in Coordination CompoundДокумент5 страницDetermination of The Percentage of Ligands in Coordination CompoundafifiОценок пока нет
- Experiment 7Документ8 страницExperiment 7Shinichi KudoОценок пока нет
- Report 4 GCДокумент26 страницReport 4 GCNurhafizah Abd JabarОценок пока нет
- Determinacion de La Vainilina Por HPLCДокумент4 страницыDeterminacion de La Vainilina Por HPLCAlfredo CruzОценок пока нет
- chm432 - 4Документ2 страницыchm432 - 4Nur AthirahОценок пока нет
- Isolation of Caffeine From A Tea BagДокумент4 страницыIsolation of Caffeine From A Tea BagohhiОценок пока нет
- Determination of caffeine in tea bag using second derivative UV spectrometryДокумент8 страницDetermination of caffeine in tea bag using second derivative UV spectrometrySuhailyShukriОценок пока нет
- The Aldol Condensation ReactionДокумент3 страницыThe Aldol Condensation ReactionJoshua CastilloОценок пока нет
- Stoichiometry Experiment AnalysisДокумент8 страницStoichiometry Experiment AnalysisHani Nadh50% (2)
- CHM510 - SpeДокумент7 страницCHM510 - SpeafifiОценок пока нет
- Experiment Baking SsodaДокумент7 страницExperiment Baking Ssodaatynzaty0% (1)
- Lab Report Chm510 Final (All Reports)Документ54 страницыLab Report Chm510 Final (All Reports)Dang HumairahОценок пока нет
- ObjectiveДокумент8 страницObjectivenaim rashidОценок пока нет
- Spectrophotometric Analysis of Transition Metal CationsДокумент5 страницSpectrophotometric Analysis of Transition Metal CationsFAtma HAnysОценок пока нет
- Chm580 Experiment 3Документ9 страницChm580 Experiment 3ohhiОценок пока нет
- CHE555 2015 Numerical Methods & Optimization AssignmentДокумент2 страницыCHE555 2015 Numerical Methods & Optimization AssignmentJaja TeukieОценок пока нет
- CSTR Design For Propylene Glycol Chemical ProductionДокумент13 страницCSTR Design For Propylene Glycol Chemical ProductionMeilani Kusuma WatiОценок пока нет
- Elc550-Exercises 4: Writing Annotated Bibliography (Benefits of Walking)Документ1 страницаElc550-Exercises 4: Writing Annotated Bibliography (Benefits of Walking)inshirahizhamОценок пока нет
- Lab CHM 420 Exp 2Документ4 страницыLab CHM 420 Exp 2nana izzОценок пока нет
- FYP Extended Proposal First DraftДокумент33 страницыFYP Extended Proposal First DraftEeHuey ChooОценок пока нет
- Chemistry of Carbonyl CompoundsДокумент28 страницChemistry of Carbonyl CompoundsRhondene WintОценок пока нет
- Nur Sarah Hannis - Exp3Документ1 страницаNur Sarah Hannis - Exp3Sarah HannisОценок пока нет
- GC Optimization Methyl EstersДокумент6 страницGC Optimization Methyl EstersaiqalОценок пока нет
- Riboflavin Concentration Fluorescence SpectroscopyДокумент5 страницRiboflavin Concentration Fluorescence SpectroscopySyahriezan HaminОценок пока нет
- Complete assessment form impacts of Covid-19Документ7 страницComplete assessment form impacts of Covid-19azim normanОценок пока нет
- Organic Chemistry II Experiment Sodium Borohydride ReductionДокумент10 страницOrganic Chemistry II Experiment Sodium Borohydride ReductionAlohaaSwezzОценок пока нет
- Exp 8Документ5 страницExp 8Lyani FaraОценок пока нет
- Experiment 3 Esterification Reactions of Vanillin: The Use of NMR To Determine A StructureДокумент1 страницаExperiment 3 Esterification Reactions of Vanillin: The Use of NMR To Determine A StructureAyish MataОценок пока нет
- Lab Report 1Документ10 страницLab Report 1sheril nur hazianiОценок пока нет
- CHM557 Exp 3Документ22 страницыCHM557 Exp 3syafОценок пока нет
- Radical PolymerizationДокумент9 страницRadical PolymerizationAtie Iekah100% (1)
- Preparation of Acetaline Notes PDFДокумент6 страницPreparation of Acetaline Notes PDFAnonymous Wwxatt3oIK100% (1)
- Covid-19 Laboratory Report Exp 5Документ7 страницCovid-19 Laboratory Report Exp 5Nasuha AriffinОценок пока нет
- LONG CAFFEINE UVLong Caffeine UvДокумент8 страницLONG CAFFEINE UVLong Caffeine UvzaОценок пока нет
- Laboratory Report CHM 457 Organic Chemistry: Universiti Teknologi Mara, Cawangan Perlis Kampus ArauДокумент4 страницыLaboratory Report CHM 457 Organic Chemistry: Universiti Teknologi Mara, Cawangan Perlis Kampus ArauNasuha AriffinОценок пока нет
- Calorimetry Lab Report: Hess's Law and MgO FormationДокумент12 страницCalorimetry Lab Report: Hess's Law and MgO Formationpufff witchesОценок пока нет
- Exp6 chm361 PDFДокумент11 страницExp6 chm361 PDFShafiqahFazyaziqahОценок пока нет
- Experiment 8: Surface Chemistry (Adsorption of Acetic Acid On Activated Carbon)Документ7 страницExperiment 8: Surface Chemistry (Adsorption of Acetic Acid On Activated Carbon)Hidayah Dayah50% (2)
- The Biochemical Roles of Transition Metals: Chm579: Advanced Inorganic ChemistryДокумент15 страницThe Biochemical Roles of Transition Metals: Chm579: Advanced Inorganic ChemistryNurul HusnaОценок пока нет
- Conclusion, Recoomendation, Reffenrence, Lab 2, CHE 485Документ2 страницыConclusion, Recoomendation, Reffenrence, Lab 2, CHE 485MOHD MU'IZZ BIN MOHD SHUKRIОценок пока нет
- Lab Report PDFДокумент21 страницаLab Report PDFLutfi Azmi0% (2)
- Iodination of HexanoneДокумент16 страницIodination of HexanonepiqotОценок пока нет
- Experiment 2 - Adsorption of Liquids Onto Solid Surfaces: TheoryДокумент3 страницыExperiment 2 - Adsorption of Liquids Onto Solid Surfaces: TheoryfrankjenОценок пока нет
- Title: Preparation of 4-MethylcyclohexeneДокумент7 страницTitle: Preparation of 4-MethylcyclohexeneNur AthirahОценок пока нет
- Lab Report Chm256 Exp 4Документ6 страницLab Report Chm256 Exp 4Miss KillerОценок пока нет
- Qualitative test carbohydrates lab reportДокумент3 страницыQualitative test carbohydrates lab reportThe seriОценок пока нет
- Influence of Air Velocity on Drying Rate of Wet SandДокумент3 страницыInfluence of Air Velocity on Drying Rate of Wet SandJohanОценок пока нет
- CMT 255 Laboratory Report: Experiment NO. Title Group Name IDДокумент9 страницCMT 255 Laboratory Report: Experiment NO. Title Group Name IDNur HismanizaОценок пока нет
- Lab Report CPP Exp 5Документ11 страницLab Report CPP Exp 5Salihah AbdullahОценок пока нет
- Introductory Titrimetric and Gravimetric Analysis: The Commonwealth and International Library: Chemistry DivisionОт EverandIntroductory Titrimetric and Gravimetric Analysis: The Commonwealth and International Library: Chemistry DivisionОценок пока нет
- Synthetic Multidentate Macrocyclic CompoundsОт EverandSynthetic Multidentate Macrocyclic CompoundsReed IzattОценок пока нет
- Thermal Decomposition AnalysisДокумент13 страницThermal Decomposition AnalysisMuhammad Danyal ShahidОценок пока нет
- Uhde Nitric Acid Process BrochureДокумент0 страницUhde Nitric Acid Process BrochureGopiОценок пока нет
- Methylamine Synthesis Over Solid ..Документ19 страницMethylamine Synthesis Over Solid ..Ravikiran Suryadevara0% (1)
- BCA AdvancedДокумент43 страницыBCA AdvancedManasvi MehtaОценок пока нет
- Lecture 1 - Enzyme & KineticsДокумент33 страницыLecture 1 - Enzyme & KineticsZeny NaranjoОценок пока нет
- Reaction Kinetics of The Catalytic Esterification of Citric Acid With EthanolДокумент8 страницReaction Kinetics of The Catalytic Esterification of Citric Acid With EthanolJason SanchezОценок пока нет
- Read 2 Optimized Biodiesel ProductionДокумент8 страницRead 2 Optimized Biodiesel ProductionKhuram MaqsoodОценок пока нет
- Polymer Degradation and StabilityДокумент10 страницPolymer Degradation and StabilityAlly HerreraОценок пока нет
- Esterifikasyon Reaktor OnemliДокумент26 страницEsterifikasyon Reaktor OnemliMehmet AydinОценок пока нет
- Chemistry - Higher Level: Pre-Leaving Certifi Cate Examination, 2014 Triailscrúdú Na Hardteistiméireachta, 2014Документ8 страницChemistry - Higher Level: Pre-Leaving Certifi Cate Examination, 2014 Triailscrúdú Na Hardteistiméireachta, 2014Diaa SaberОценок пока нет
- How Electrochemical Gas Sensors WorkДокумент4 страницыHow Electrochemical Gas Sensors Workagsan.algabh2718Оценок пока нет
- Photo DegradationДокумент24 страницыPhoto DegradationMaica Caguiat100% (1)
- United States Patent (19) 11 Patent Number: 5,935,415Документ12 страницUnited States Patent (19) 11 Patent Number: 5,935,415xyz7890Оценок пока нет
- PDFДокумент91 страницаPDFSatyam KumarОценок пока нет
- New Journal of Chemistry January 2009aДокумент222 страницыNew Journal of Chemistry January 2009aNguyên NhãОценок пока нет
- Introduction To Inherently Safer Design: Prepared For Safety and Chemical Engineering Education (SACHE) byДокумент71 страницаIntroduction To Inherently Safer Design: Prepared For Safety and Chemical Engineering Education (SACHE) bySebastian iacopiОценок пока нет
- What Affects Enzyme Activity-FinalДокумент3 страницыWhat Affects Enzyme Activity-FinalCho Ah YoungОценок пока нет
- 11 Chapter Reaction Kinetics Text Book ExerciseДокумент14 страниц11 Chapter Reaction Kinetics Text Book ExerciseSajid AzeemОценок пока нет
- Kinetics of Ester HydrolysisДокумент24 страницыKinetics of Ester HydrolysisNick OnuskaОценок пока нет
- ACS NE Book Proof01-1Документ108 страницACS NE Book Proof01-1d anjilappaОценок пока нет
- Lithium-Imide Ammonia Decomp Catalysts DavidДокумент9 страницLithium-Imide Ammonia Decomp Catalysts Davidkwayneolson6081Оценок пока нет
- Unit Operations and Unit ProcessesДокумент5 страницUnit Operations and Unit ProcesseslaurenОценок пока нет
- Supramolecular Catalysis PDFДокумент9 страницSupramolecular Catalysis PDFmradu1Оценок пока нет
- Topic - 17 - Past - Papers MarkschemeДокумент10 страницTopic - 17 - Past - Papers MarkschemeMahmoud Sameh-Abdel-LateefОценок пока нет
- Lecture 14 Air PollutionДокумент45 страницLecture 14 Air PollutionAyushi AggarwalОценок пока нет
- Zeolites in Petroleum RefiningДокумент6 страницZeolites in Petroleum RefiningAlina SmochinaОценок пока нет
- Chapter 4 - Enzymes & MetabolismДокумент4 страницыChapter 4 - Enzymes & MetabolismGrace LeungОценок пока нет
- Exhaust SystemДокумент16 страницExhaust SystemArvind KatyayanОценок пока нет
- Amazon Cafe.9015.1474873367Документ14 страницAmazon Cafe.9015.1474873367AsmZziz OoОценок пока нет
- Jeyalakshmi Thesis 2Документ271 страницаJeyalakshmi Thesis 2Farah Talib Al-sudaniОценок пока нет
- Prereformer CatalystДокумент20 страницPrereformer CatalystFanni Boying100% (3)