Вам также может понравиться
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (119)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2219)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- Pharm MnemonicsДокумент33 страницыPharm MnemonicsThomson George75% (4)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (890)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Hospital Job DescriptionsДокумент48 страницHospital Job DescriptionsLoresita Amoranto Rebong RNОценок пока нет
- Pediatric Voice PresentationДокумент16 страницPediatric Voice Presentationapi-254429093Оценок пока нет
- Part and Mold Design GuideДокумент170 страницPart and Mold Design GuideminhtintinОценок пока нет
- Mini Question Bank - Vety Sci - For Students PDFДокумент106 страницMini Question Bank - Vety Sci - For Students PDFRakesh Prajapati100% (3)
- Ten Days in A Mad-House by Nellie BlyДокумент122 страницыTen Days in A Mad-House by Nellie BlyCharmaineTanti100% (1)
- NCP CompilationДокумент11 страницNCP CompilationRene John FranciscoОценок пока нет
- The Hypothesis and Assumptions of The StudyДокумент12 страницThe Hypothesis and Assumptions of The StudyMaria Arlene67% (3)
- Uttar Pradesh Empanelment List for Minilap 2018-19Документ19 страницUttar Pradesh Empanelment List for Minilap 2018-19Vijay KumarОценок пока нет
- Facs ProtocolДокумент7 страницFacs ProtocolmisterxОценок пока нет
- BT 2019Документ13 страницBT 2019biotech_vidhyaОценок пока нет
- Stripping For ReprobingДокумент2 страницыStripping For ReprobingStella SalvatoreОценок пока нет
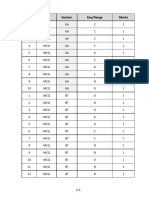
- Q.No. Type Section Key/Range MarksДокумент3 страницыQ.No. Type Section Key/Range Marksbiotech_vidhyaОценок пока нет
- Stripping For ReprobingДокумент2 страницыStripping For ReprobingStella SalvatoreОценок пока нет
- Polymerase Chain Reaction (PCR)Документ3 страницыPolymerase Chain Reaction (PCR)biotech_vidhyaОценок пока нет
- Brad FordДокумент12 страницBrad FordQi ChaoОценок пока нет
- SDS PageДокумент2 страницыSDS Pagebiotech_vidhyaОценок пока нет
- Buffer Preparation Guide for DNA/Protein Work (Shi LabДокумент6 страницBuffer Preparation Guide for DNA/Protein Work (Shi Labbiotech_vidhyaОценок пока нет
- Troubleshooting SDS-PAGE 1Документ3 страницыTroubleshooting SDS-PAGE 1biotech_vidhyaОценок пока нет
- Polymerasen GuideДокумент16 страницPolymerasen Guidebiotech_vidhyaОценок пока нет
- Components Reaction MixtureДокумент3 страницыComponents Reaction Mixturebiotech_vidhyaОценок пока нет
- Buffer Preparation Guide for DNA/Protein Work (Shi LabДокумент6 страницBuffer Preparation Guide for DNA/Protein Work (Shi Labbiotech_vidhyaОценок пока нет
- TNPSC Group 1 Prelim Book List PDFДокумент2 страницыTNPSC Group 1 Prelim Book List PDFbiotech_vidhyaОценок пока нет
- Whole Cell ExtractДокумент1 страницаWhole Cell Extractbiotech_vidhyaОценок пока нет
- TNPSC Group 1 Prelim Book List PDFДокумент2 страницыTNPSC Group 1 Prelim Book List PDFbiotech_vidhyaОценок пока нет
- Befcv List PDFДокумент22 страницыBefcv List PDFbiotech_vidhyaОценок пока нет
- Nuclear ExtractsДокумент2 страницыNuclear Extractsbiotech_vidhyaОценок пока нет
- Qpaper PondyДокумент21 страницаQpaper Pondybiotech_vidhyaОценок пока нет
- Ies 17 Set A Me Q AДокумент67 страницIes 17 Set A Me Q Abiotech_vidhyaОценок пока нет
- TNPSC Group 1 Prelim Book List PDFДокумент2 страницыTNPSC Group 1 Prelim Book List PDFbiotech_vidhyaОценок пока нет
- A.E. (Mechanical Engineering I) 2007Документ24 страницыA.E. (Mechanical Engineering I) 2007Mukesh KumarОценок пока нет
- Qpaper PondyДокумент21 страницаQpaper Pondybiotech_vidhyaОценок пока нет
- ESE 2017 Mechanical Engineering Prelims Exam Detailed SolutionДокумент52 страницыESE 2017 Mechanical Engineering Prelims Exam Detailed SolutionpataОценок пока нет
- Img Word-To PDFДокумент3 страницыImg Word-To PDFbiotech_vidhyaОценок пока нет
- Mechanical Engineering Code No. 14: Combined Competitive (Preliminary) Examination, 2010Документ20 страницMechanical Engineering Code No. 14: Combined Competitive (Preliminary) Examination, 2010biotech_vidhyaОценок пока нет
- TDC 41597 A (Mechanical Engg.) - 2012Документ20 страницTDC 41597 A (Mechanical Engg.) - 2012biotech_vidhyaОценок пока нет
- Recruitment RulesДокумент5 страницRecruitment Rulesbiotech_vidhyaОценок пока нет
- 1 TolerancesДокумент1 страница1 Tolerancesbiotech_vidhyaОценок пока нет
- Assessment Diagnosis Planning Interventions Rationale Evaluation Subjective: "Maglisod Man Kog Short Term: Independent: - Establish RapportДокумент3 страницыAssessment Diagnosis Planning Interventions Rationale Evaluation Subjective: "Maglisod Man Kog Short Term: Independent: - Establish RapportSergi Lee OrateОценок пока нет
- Penjualan Total Juli 19Документ266 страницPenjualan Total Juli 19wartiniОценок пока нет
- Mayo HospitalДокумент1 страницаMayo HospitalTooba SaeedОценок пока нет
- PCRL 627: Clinical Microbiology and Immunology 2016: Email: Shilling@uic - EduДокумент45 страницPCRL 627: Clinical Microbiology and Immunology 2016: Email: Shilling@uic - Edui24youОценок пока нет
- Mapping Aqsha 1: Heart Failure, Esophageal Cancer, and MoreДокумент7 страницMapping Aqsha 1: Heart Failure, Esophageal Cancer, and MorePutri Rahmadhani Ngakpaniklage AsdsОценок пока нет
- A Study of Dissolution Enhancement and Invitro Evaluation of RoxithromycinДокумент6 страницA Study of Dissolution Enhancement and Invitro Evaluation of Roxithromycinanto_pharma7784Оценок пока нет
- Bell's PalsyДокумент12 страницBell's PalsyMercy nafulaОценок пока нет
- 2001 PP No 22Документ10 страниц2001 PP No 22tarОценок пока нет
- Meier Et Al, 1996Документ4 страницыMeier Et Al, 1996boni_sebayangОценок пока нет
- Partners Case CCMNДокумент4 страницыPartners Case CCMNapi-314349758Оценок пока нет
- 12-Channel ECG SpecsДокумент5 страниц12-Channel ECG SpecsNai PaniaiОценок пока нет
- Klinefelter SyndromeДокумент6 страницKlinefelter Syndromemeeeenon100% (1)
- BUFFER STOK SO TGL 27des2021Документ3 страницыBUFFER STOK SO TGL 27des2021Putu NindyaОценок пока нет
- Showtime - Smilf 1x01 (Pilot)Документ36 страницShowtime - Smilf 1x01 (Pilot)Vishnu SinhaОценок пока нет
- Comparing MUAC and weight-for-height to diagnose SAMДокумент30 страницComparing MUAC and weight-for-height to diagnose SAMsidhant pundhirОценок пока нет
- EORTC QLQ-C30 Into Indonesian VersionДокумент11 страницEORTC QLQ-C30 Into Indonesian VersionHendryОценок пока нет
- 2017 Functional Anatomy Test QuestionsДокумент10 страниц2017 Functional Anatomy Test QuestionsmarcusОценок пока нет
- Aeromedical Transport TPДокумент8 страницAeromedical Transport TPgalib20Оценок пока нет
- Aj. Chulaporn-Bosutinib-Search Engine and Study EndpointsДокумент5 страницAj. Chulaporn-Bosutinib-Search Engine and Study EndpointsCalm Phurit SenachaiОценок пока нет
- Articles - Is SX Addiction Real - Help4SexualAddictionДокумент2 страницыArticles - Is SX Addiction Real - Help4SexualAddictionanacconzattiОценок пока нет
- NCP OrthoДокумент2 страницыNCP OrthoJeyser T. GamutiaОценок пока нет
- Spatial Analysis of Pulmonary TB Distribution and Risk FactorsДокумент7 страницSpatial Analysis of Pulmonary TB Distribution and Risk FactorsAhmad RivaiОценок пока нет