Вам также может понравиться
- LM338TДокумент17 страницLM338Talvaldez035981Оценок пока нет
- 1993 Voigt - Hydroxylation of The Native Brassinosteroids 24-Epicastasterone and 24-Epibrassinolide by The Fungus Cunninghamella Echinulata PDFДокумент4 страницы1993 Voigt - Hydroxylation of The Native Brassinosteroids 24-Epicastasterone and 24-Epibrassinolide by The Fungus Cunninghamella Echinulata PDFHernán AstudilloОценок пока нет
- 1993 Ando - 13C NMR Assignments of Brassinosteroids by 2-Dimensional Techniques PDFДокумент6 страниц1993 Ando - 13C NMR Assignments of Brassinosteroids by 2-Dimensional Techniques PDFHernán AstudilloОценок пока нет
- 1987 Mayr - Acid - and Base-Catalyzed Ring-Opening Reactions or A Sterically Hindered Epoxide PDFДокумент3 страницы1987 Mayr - Acid - and Base-Catalyzed Ring-Opening Reactions or A Sterically Hindered Epoxide PDFHernán AstudilloОценок пока нет
- A New Type of Insulated Molecular Wire - A Rotaxane DerivedДокумент3 страницыA New Type of Insulated Molecular Wire - A Rotaxane DerivedHernán AstudilloОценок пока нет
- Adv Calculus and AnalysisДокумент110 страницAdv Calculus and AnalysisAshok PhoenixОценок пока нет
- Selection of Suitable Propagation Method PDFДокумент8 страницSelection of Suitable Propagation Method PDFHernán AstudilloОценок пока нет
- Conceptos de Electroquímica - Microbalanza de CuarzoДокумент25 страницConceptos de Electroquímica - Microbalanza de CuarzoHernán AstudilloОценок пока нет
- 2,4-Dinitrophenol: A Threat To Chinese Body-Conscious GroupsДокумент3 страницы2,4-Dinitrophenol: A Threat To Chinese Body-Conscious GroupsHernán AstudilloОценок пока нет
- Origin of Enantioselectivity in The Jacobse Epoxidation of OlefinsДокумент4 страницыOrigin of Enantioselectivity in The Jacobse Epoxidation of OlefinsHernán AstudilloОценок пока нет
- 44th International Chemistry Olympiad Preparatory Problems Dec 2-2Документ75 страниц44th International Chemistry Olympiad Preparatory Problems Dec 2-2lightningfaxОценок пока нет
- Contribuciones de La Electrosíntesis Orgánica A La Química VerdeДокумент21 страницаContribuciones de La Electrosíntesis Orgánica A La Química VerdeHernán AstudilloОценок пока нет
- TLC 555Документ25 страницTLC 555Hernán AstudilloОценок пока нет
- Carbohidratos - Determinación - Use of The Alditol Acetate Derivatisation For The Analysis of Reducing Sugars in Potato TubersДокумент5 страницCarbohidratos - Determinación - Use of The Alditol Acetate Derivatisation For The Analysis of Reducing Sugars in Potato TubersHernán AstudilloОценок пока нет
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5782)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (890)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (265)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (119)
- Andrew - HBX 6516DS VTM - HBX 6516DS A1MДокумент3 страницыAndrew - HBX 6516DS VTM - HBX 6516DS A1MVu Minh DuongОценок пока нет
- 01.1. Big BangДокумент17 страниц01.1. Big BangLord William PacuribОценок пока нет
- Cume213 L2Документ80 страницCume213 L2irvinvongiОценок пока нет
- Robust Wax Deposition Modeling Incorporating NonДокумент4 страницыRobust Wax Deposition Modeling Incorporating Nonabhay.singhОценок пока нет
- QI Lecture NotesДокумент83 страницыQI Lecture NotesMiguel A. BastarracheaОценок пока нет
- The Arrohan: Chemistry Test Class-XI Topic-Atomic Structure & Chemical Bonding F.M-40Документ2 страницыThe Arrohan: Chemistry Test Class-XI Topic-Atomic Structure & Chemical Bonding F.M-40AdityaОценок пока нет
- Ultrasonic MotorДокумент19 страницUltrasonic MotorKallol_Bera221Оценок пока нет
- Discretization Considerations in Moving Load Finite Element Beam ModelsДокумент16 страницDiscretization Considerations in Moving Load Finite Element Beam ModelsEugene CommerellОценок пока нет
- 2.2.7. Optical RotationДокумент2 страницы2.2.7. Optical RotationTrung Dương ĐứcОценок пока нет
- Mod 1 Atoms Bonds RxnstestansДокумент2 страницыMod 1 Atoms Bonds Rxnstestansrichardgharexd1Оценок пока нет
- Rizal General Chemistry 2 q3 Slm4Документ12 страницRizal General Chemistry 2 q3 Slm4Darlene OpeñaОценок пока нет
- GRHW 1Документ2 страницыGRHW 1Kastalia Delaporta0% (1)
- MDI PurityДокумент6 страницMDI PuritycgrigorasОценок пока нет
- Formation of iodobenzene from arenediazonium salts and iodide ionsДокумент3 страницыFormation of iodobenzene from arenediazonium salts and iodide ionsCamiloVerdugoОценок пока нет
- Gauss's Law and DivergenceДокумент11 страницGauss's Law and DivergenceErxDОценок пока нет
- Shigley Mohr Circle NotesДокумент24 страницыShigley Mohr Circle NotesPaul WallsОценок пока нет
- Triple Point Plot: Melting FreezingДокумент16 страницTriple Point Plot: Melting FreezingJorgeОценок пока нет
- ECLIPSE 50i ECLIPSE 50i 2033.05 - Microscope - Lab, - AccessoriesДокумент5 страницECLIPSE 50i ECLIPSE 50i 2033.05 - Microscope - Lab, - Accessoriesluroguita-1Оценок пока нет
- Pure Mathematics A-Level Paper 2: 2002-AL P MathДокумент26 страницPure Mathematics A-Level Paper 2: 2002-AL P MathwltfОценок пока нет
- Mimw With CoverДокумент13 страницMimw With Coverank123qwerОценок пока нет
- ME 395 12 Solutions #5Документ5 страницME 395 12 Solutions #5me395100% (2)
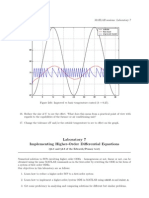
- MATLAB sessions: Laboratory 7 implementing higher-order differential equationsДокумент7 страницMATLAB sessions: Laboratory 7 implementing higher-order differential equationsjlbalbОценок пока нет
- Mordern ABC Physics PDFДокумент250 страницMordern ABC Physics PDFLakky Srinu P90% (10)
- PhysicsLAB - Period of A PendulumДокумент3 страницыPhysicsLAB - Period of A PendulumCitizen Kwadwo AnsongОценок пока нет
- COADE Friction StiffnessДокумент1 страницаCOADE Friction Stiffnessniteshk_45Оценок пока нет
- Le ChatelierДокумент12 страницLe ChatelierYan LaksanaОценок пока нет
- Iso Astm 52701-13 PDFДокумент10 страницIso Astm 52701-13 PDFAhmed LabibОценок пока нет
- Instruction Bulletin Dry-Type Transformers 600 Volts and BelowДокумент24 страницыInstruction Bulletin Dry-Type Transformers 600 Volts and BelowMiguel Blasco Martínez0% (1)
- Fick's Law of Diffusion: Imaicela Gloria, Calderón Luis, Romero Henry, Criollo Erick, Trujillo WillamДокумент5 страницFick's Law of Diffusion: Imaicela Gloria, Calderón Luis, Romero Henry, Criollo Erick, Trujillo WillamLuis Calderon SalasОценок пока нет
- General Kinetics of RotationДокумент9 страницGeneral Kinetics of Rotationdenver costalesОценок пока нет