Вам также может понравиться
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (345)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- Wrestling Strength and ConditioningДокумент12 страницWrestling Strength and ConditioningTintin BilatbatОценок пока нет
- CFM Tutorial 5Документ26 страницCFM Tutorial 5Nithin Yadav0% (1)
- Nutritional Classification of BacteriaДокумент7 страницNutritional Classification of BacteriaRalphpinno SanchezОценок пока нет
- Medical Imaging WebquestДокумент8 страницMedical Imaging Webquestapi-262193618Оценок пока нет
- Treeleaf Basic Zazen InstructionsДокумент16 страницTreeleaf Basic Zazen InstructionsFaisal sarhiОценок пока нет
- TOCДокумент14 страницTOCAthirah HattaОценок пока нет
- Publication Edition 2020Документ230 страницPublication Edition 2020Mech Dept GMITОценок пока нет
- Lesson 2.1 Earth As The Only Habitable PlanetДокумент37 страницLesson 2.1 Earth As The Only Habitable Planetrosie sialanaОценок пока нет
- Specification and Maintenance Manual: Sequence Flashing Lights (SFL)Документ13 страницSpecification and Maintenance Manual: Sequence Flashing Lights (SFL)Javier Eduardo Alzate BogotaОценок пока нет
- NEWS BD RAE Letter of Intent-Press-release1Документ2 страницыNEWS BD RAE Letter of Intent-Press-release1Anthony D.Оценок пока нет
- Nomenclature Practice ProblemsДокумент4 страницыNomenclature Practice ProblemsMax DavidsonОценок пока нет
- Thai Cuisine: Reporters: Bantayan, Kenneth Samejon, Clarish Lovely Relevo, Mary GraceДокумент47 страницThai Cuisine: Reporters: Bantayan, Kenneth Samejon, Clarish Lovely Relevo, Mary Gracemiralona relevoОценок пока нет
- High Prices Most Unique ProductsДокумент1 страницаHigh Prices Most Unique ProductsJusteen BalcortaОценок пока нет
- G1 CurvedДокумент16 страницG1 CurvedElbert Ryan OcampoОценок пока нет
- Membrane AutopsyДокумент2 страницыMembrane AutopsyBiljana TausanovicОценок пока нет
- The Prosecution of Kim Jong Il - Accountability in A Post 9-11 WorldДокумент21 страницаThe Prosecution of Kim Jong Il - Accountability in A Post 9-11 WorldimpunitywatchОценок пока нет
- Achai, Sydney Jill S. GE 15 - SIM - ULOcДокумент13 страницAchai, Sydney Jill S. GE 15 - SIM - ULOcSydney AchaiОценок пока нет
- Rec Site Visit Report 1Документ6 страницRec Site Visit Report 1api-474887597Оценок пока нет
- GST15!16!17-Bad Debt Relief RecoverДокумент10 страницGST15!16!17-Bad Debt Relief RecoverDaud Farook IIОценок пока нет
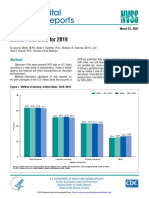
- National Vital Statistics Reports: Births: Final Data For 2019Документ51 страницаNational Vital Statistics Reports: Births: Final Data For 2019Simon ReichОценок пока нет
- Leadership Roles and Management Functions in Nursing Theory and ApplicationДокумент2 страницыLeadership Roles and Management Functions in Nursing Theory and Applicationivan0% (3)
- Chapter 1 Section 6 Spoon Feeding BasicsДокумент9 страницChapter 1 Section 6 Spoon Feeding Basicskenneth mayaoОценок пока нет
- Low Cholesterol DietДокумент10 страницLow Cholesterol Dietkevintotz73Оценок пока нет
- Kidney Diet DelightsДокумент20 страницKidney Diet DelightsArturo Treviño MedinaОценок пока нет
- Nama Anggota: Dede Wiyanto Endri Murni Hati Rukhi Hasibah Tugas: Bahasa Inggris (Narrative Text)Документ3 страницыNama Anggota: Dede Wiyanto Endri Murni Hati Rukhi Hasibah Tugas: Bahasa Inggris (Narrative Text)Wiyan Alwaysfans CheLseaОценок пока нет
- Showalter Female MaladyДокумент13 страницShowalter Female MaladyKevin Sebastian Patarroyo GalindoОценок пока нет
- Brooklyn Hops BreweryДокумент24 страницыBrooklyn Hops BrewerynyairsunsetОценок пока нет
- Problem Set in Power System 2Документ3 страницыProblem Set in Power System 2Andrew AlterОценок пока нет
- Aahaa Puttu Flour ProjectДокумент53 страницыAahaa Puttu Flour ProjectApple ComputersОценок пока нет
- Field Study 1-Act 5.1Документ5 страницField Study 1-Act 5.1Mariya QuedzОценок пока нет