Вам также может понравиться
- Isolation and Characterization of DNAДокумент75 страницIsolation and Characterization of DNANathaniel CastasusОценок пока нет
- Molecular Biology Lab Genomic DNA ExtractionДокумент19 страницMolecular Biology Lab Genomic DNA ExtractionVineet Kumar ThakurОценок пока нет
- Recombinant DNA TechnologyДокумент26 страницRecombinant DNA TechnologyManikandan VpОценок пока нет
- Heredity of Dna: - Prokaryotes - EukaryotesДокумент22 страницыHeredity of Dna: - Prokaryotes - EukaryotesCharles RajОценок пока нет
- Biochemical and Molecular Genetics: Bche 545/molb 545Документ54 страницыBiochemical and Molecular Genetics: Bche 545/molb 545naveenmi2Оценок пока нет
- Southern BlottingДокумент23 страницыSouthern BlottingAbdul BasitОценок пока нет
- Nucleic-Acid Isolation Methods: Michael T Madziva, PHDДокумент33 страницыNucleic-Acid Isolation Methods: Michael T Madziva, PHDmibritow23Оценок пока нет
- Molecular Cloning: Dr. Robin Herlands Genetics, Biology 300, Nevada State CollegeДокумент56 страницMolecular Cloning: Dr. Robin Herlands Genetics, Biology 300, Nevada State CollegeSamir PatelОценок пока нет
- 18 - Flow Cytometry and Introduction To Molecular PathologyДокумент11 страниц18 - Flow Cytometry and Introduction To Molecular Pathologyhamadadodo7Оценок пока нет
- L4 Dna Preparation MethodsДокумент24 страницыL4 Dna Preparation MethodsSandra MwendeОценок пока нет
- DNA ExtractionДокумент56 страницDNA ExtractionZain YaqoobОценок пока нет
- Js 190 - Dna Extraction MethodsДокумент56 страницJs 190 - Dna Extraction MethodsKelvin LeongОценок пока нет
- I Recombinant DNA TechnologyДокумент37 страницI Recombinant DNA TechnologyGundeep BrarОценок пока нет
- DNA Extraction MethodsДокумент22 страницыDNA Extraction MethodsGarshel KellyОценок пока нет
- DNA LibraryДокумент73 страницыDNA LibraryDGA GAMINGОценок пока нет
- Biochem Lec 3Документ97 страницBiochem Lec 3yashika gargОценок пока нет
- Biol3451 Ch19 LectДокумент37 страницBiol3451 Ch19 LectmaniiiiiiiiОценок пока нет
- INTRODUCTION Isolation of Plant DNAДокумент23 страницыINTRODUCTION Isolation of Plant DNAkhushbujain7992Оценок пока нет
- Nucleic Acid Biotechnology Techniques: Mary K. Campbell Shawn O. FarrellДокумент40 страницNucleic Acid Biotechnology Techniques: Mary K. Campbell Shawn O. Farrellleng cuetoОценок пока нет
- DNA Extraction From MeatДокумент25 страницDNA Extraction From Meatnoor saqibОценок пока нет
- Blotting SystemsДокумент61 страницаBlotting SystemsAdebisi OluwatomiwaОценок пока нет
- Lec 5 DNA Extraction and PCRДокумент25 страницLec 5 DNA Extraction and PCRSaif MohamedОценок пока нет
- Chapter 3: Nucleic Acids: Presented by Dr. Shazzad Hosain Asst. Prof. EECS, NSUДокумент75 страницChapter 3: Nucleic Acids: Presented by Dr. Shazzad Hosain Asst. Prof. EECS, NSUAlimushwan AdnanОценок пока нет
- 618 Restriction Enzymes and VectorsДокумент25 страниц618 Restriction Enzymes and Vectorsakshif KhanОценок пока нет
- BOTA 801 Presentation 2Документ50 страницBOTA 801 Presentation 2jackson kesseОценок пока нет
- FISH GENETICS TechniquesДокумент22 страницыFISH GENETICS TechniquesHanna Beth Butiong TandayagОценок пока нет
- P. Bioteknologi - Teknologi Rekombinan DNA - 2022Документ41 страницаP. Bioteknologi - Teknologi Rekombinan DNA - 2022Bruneitabba Billal IrawanОценок пока нет
- Cloning and Re Comb in Ant DnaДокумент38 страницCloning and Re Comb in Ant DnaMak ItiОценок пока нет
- Chapter 6Документ17 страницChapter 6Lindsay OttoОценок пока нет
- 12 Chap11 Biotech 1597571962Документ67 страниц12 Chap11 Biotech 1597571962Maheswari RajnarayananОценок пока нет
- BT 403+mod III+NKJ+Lecture+ 2 DNA+Isolation 2011Документ8 страницBT 403+mod III+NKJ+Lecture+ 2 DNA+Isolation 2011tulibhawalОценок пока нет
- Genomic DNA ExtractionДокумент18 страницGenomic DNA Extractionaenarae01Оценок пока нет
- DNA Isolation, PCR & Sequencing: Dr. Dr. Mgs. Irsan Saleh, M.BiomedДокумент22 страницыDNA Isolation, PCR & Sequencing: Dr. Dr. Mgs. Irsan Saleh, M.BiomedIqbal AlRabbОценок пока нет
- Recombinant DNA Technology LectureДокумент50 страницRecombinant DNA Technology LectureJean Carmelette BalalloОценок пока нет
- DNA Isolation: Doc. Ruslan KalendarДокумент20 страницDNA Isolation: Doc. Ruslan KalendarAswina NadiaОценок пока нет
- Principles of Dna Isolation & PurificationДокумент47 страницPrinciples of Dna Isolation & PurificationYeyen Musaini100% (1)
- Restriction Analysis Lecture Bioinformatics UaarДокумент18 страницRestriction Analysis Lecture Bioinformatics Uaarahmed fouadОценок пока нет
- DNA Extraction OverviewДокумент14 страницDNA Extraction OverviewNicholas SoОценок пока нет
- DNA Isolation VIДокумент68 страницDNA Isolation VIMohamed AliОценок пока нет
- 3.0 Basic Molecular TechniquesДокумент70 страниц3.0 Basic Molecular Techniquesvivian kiuОценок пока нет
- Genetics Lecture 2 - DNA and Chromosome Structure PDFДокумент58 страницGenetics Lecture 2 - DNA and Chromosome Structure PDFMarlee SandersonОценок пока нет
- 14th WeekДокумент21 страница14th WeekMoonHoLeeОценок пока нет
- Restriction Enzyme DigestionДокумент8 страницRestriction Enzyme DigestionSaba IkhlaqОценок пока нет
- DNA Extraction From E. ColiДокумент33 страницыDNA Extraction From E. ColiAlaa Al AdawiОценок пока нет
- Recombinant DNA Technology: Common General Cloning StrategyДокумент42 страницыRecombinant DNA Technology: Common General Cloning Strategyhugo200887Оценок пока нет
- Loc 6Документ47 страницLoc 6Patricia Jayshree Samuel JacobОценок пока нет
- T2 Syllabus Revision ClassДокумент73 страницыT2 Syllabus Revision ClassOhhh OkayОценок пока нет
- Lab Techniques: James Chappell & Cheuk Ka TongДокумент31 страницаLab Techniques: James Chappell & Cheuk Ka TongEliza LindaОценок пока нет
- DNA Isolation, PCR & Sequencing: Dr. Dr. Mgs. Irsan Saleh, M.BiomedДокумент22 страницыDNA Isolation, PCR & Sequencing: Dr. Dr. Mgs. Irsan Saleh, M.BiomedUmiieg MiansyahОценок пока нет
- Nucleic Acid Dna and Rna Isolation: Molecular DiagnosticsДокумент2 страницыNucleic Acid Dna and Rna Isolation: Molecular DiagnosticsPaul LesterОценок пока нет
- Enzyeme Restriction DigestionДокумент18 страницEnzyeme Restriction DigestionEndang PurwantiОценок пока нет
- GCT 1Документ58 страницGCT 1Wajid KhattakОценок пока нет
- Isolasi DNA WatsonДокумент59 страницIsolasi DNA WatsonHimmatul UlyaОценок пока нет
- Lecture 6-Gene CloningДокумент43 страницыLecture 6-Gene CloningNurfarahain ZolkeflyОценок пока нет
- Principles of Molecular BiologyДокумент39 страницPrinciples of Molecular BiologyNando NanginОценок пока нет
- Genetic Engineering: Aliyu, HabibuДокумент32 страницыGenetic Engineering: Aliyu, HabibuAkinsanmi TaiwoОценок пока нет
- Recombinant DNA Technology: Basic Laboratory Techniques & EquipmentДокумент35 страницRecombinant DNA Technology: Basic Laboratory Techniques & EquipmentSylviaОценок пока нет
- RDNA Lect Lecture Notes Mid Sem BTech IDDДокумент146 страницRDNA Lect Lecture Notes Mid Sem BTech IDDarpna sjsОценок пока нет
- Transformation ConfirmationДокумент5 страницTransformation ConfirmationMuhammad MoeezОценок пока нет
- Cleft Palate JournalДокумент5 страницCleft Palate JournalJonathan ChanОценок пока нет
- 9 Rules of Inference 10 Rules of Replacement Symbols in LogicДокумент2 страницы9 Rules of Inference 10 Rules of Replacement Symbols in LogicJonathan ChanОценок пока нет
- A Review of The Economic Evidence of Typhoid Fever and Typhoid VaccinesДокумент13 страницA Review of The Economic Evidence of Typhoid Fever and Typhoid VaccinesJonathan ChanОценок пока нет
- 1245 FullДокумент15 страниц1245 FullJonathan ChanОценок пока нет
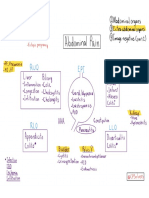
- WP Contentuploads201901Abdominal Pain Overview PDFДокумент1 страницаWP Contentuploads201901Abdominal Pain Overview PDFJonathan ChanОценок пока нет
- 18 Aus-French Armed ForcesДокумент9 страниц18 Aus-French Armed ForcesJonathan ChanОценок пока нет
- 19 Aus-Wagner Et AlДокумент8 страниц19 Aus-Wagner Et AlJonathan ChanОценок пока нет
- May 8, Tuesday May 10, Thursday 12:45 PM Onwards: TestДокумент7 страницMay 8, Tuesday May 10, Thursday 12:45 PM Onwards: TestJonathan ChanОценок пока нет
- nbt1004 1315 PDFДокумент2 страницыnbt1004 1315 PDFJonathan ChanОценок пока нет
- Student GuideДокумент1 страницаStudent GuideJonathan ChanОценок пока нет
- Henderson-Hasselbalch: Suppose Naoac Is Added To The Solution in Example 1, What Will Happen?Документ1 страницаHenderson-Hasselbalch: Suppose Naoac Is Added To The Solution in Example 1, What Will Happen?Jonathan ChanОценок пока нет
- Henderson-Hasselbalch: Suppose Naoac Is Added To The Solution in Example 1, What Will Happen?Документ1 страницаHenderson-Hasselbalch: Suppose Naoac Is Added To The Solution in Example 1, What Will Happen?Jonathan ChanОценок пока нет
- Molecular Dynamics and Drug Discovery (2016) - NotesДокумент1 страницаMolecular Dynamics and Drug Discovery (2016) - NotesJonathan ChanОценок пока нет
- Fascial PlanesДокумент10 страницFascial PlanesJonathan ChanОценок пока нет
- Geol 11 Sedimentary EnvironmentsДокумент5 страницGeol 11 Sedimentary EnvironmentsJonathan ChanОценок пока нет
- Molecular Dynamics: An in Silico Analysis of JD and Its MutantДокумент7 страницMolecular Dynamics: An in Silico Analysis of JD and Its MutantJonathan ChanОценок пока нет
- Student Participation Agreement (SPA)Документ2 страницыStudent Participation Agreement (SPA)Jonathan ChanОценок пока нет
- Molecular Dynamics of Drug ResistanceДокумент10 страницMolecular Dynamics of Drug ResistanceJonathan ChanОценок пока нет
- Arterial System LIДокумент1 страницаArterial System LIJonathan ChanОценок пока нет
- PI 100 Social ClassesДокумент2 страницыPI 100 Social ClassesJonathan ChanОценок пока нет
- LSM1102 - Alkaline Lysis Original ArticleДокумент11 страницLSM1102 - Alkaline Lysis Original Articlegivena2ndchanceОценок пока нет
- Proteins and Linguistics HandoutДокумент2 страницыProteins and Linguistics HandoutJonathan ChanОценок пока нет
- Course Syllabus Biology 180 Statistical Methods in BiologyДокумент3 страницыCourse Syllabus Biology 180 Statistical Methods in BiologyJonathan ChanОценок пока нет
- Army Pocket Guide: Physical TrainingДокумент140 страницArmy Pocket Guide: Physical Trainingsaravanan_techОценок пока нет
- Bio 12 3rd Exam TranscriptДокумент6 страницBio 12 3rd Exam TranscriptJonathan ChanОценок пока нет
- UP Diliman Acad Calendar 2014-2015Документ1 страницаUP Diliman Acad Calendar 2014-2015FMDCОценок пока нет
- Bio 12 4th Exam TranscriptДокумент7 страницBio 12 4th Exam TranscriptJonathan ChanОценок пока нет
- Plant HormonesДокумент5 страницPlant HormonesJonathan ChanОценок пока нет
- Bio 12 Special Project Capsule ProposalДокумент2 страницыBio 12 Special Project Capsule ProposalJonathan ChanОценок пока нет
- Amgen Inc. v. F. Hoffmann-LaRoche LTD Et Al - Document No. 1142Документ16 страницAmgen Inc. v. F. Hoffmann-LaRoche LTD Et Al - Document No. 1142Justia.comОценок пока нет
- Ebook PDF Concepts of Genetics Global Edition 12th Edition PDFДокумент41 страницаEbook PDF Concepts of Genetics Global Edition 12th Edition PDFamy.martin707100% (38)
- POGIL Central Dogma PDFДокумент2 страницыPOGIL Central Dogma PDFZoe101Оценок пока нет
- Transcription Prokaryotes 2012-cДокумент27 страницTranscription Prokaryotes 2012-cAnupama PatiОценок пока нет
- Bioknowledgy Quick Quiz On Transcription & Gene Expression (7.2)Документ2 страницыBioknowledgy Quick Quiz On Transcription & Gene Expression (7.2)priyaОценок пока нет
- Probabilistic Epigenesis: Gilbert GottliebДокумент11 страницProbabilistic Epigenesis: Gilbert Gottliebgrupo de investigacion dneuropsy fac.saludОценок пока нет
- The Birth and Death of Proteins: Some Key ConceptsДокумент2 страницыThe Birth and Death of Proteins: Some Key ConceptsLuke ShantiОценок пока нет
- No COVID 19 Transmission in Semen Observed in Initial Work 1587026939Документ11 страницNo COVID 19 Transmission in Semen Observed in Initial Work 1587026939Ioana Adina RugescuОценок пока нет
- Craniofacial Biology - Gene Expression During Tooth DevelopmentДокумент3 страницыCraniofacial Biology - Gene Expression During Tooth DevelopmentzeljkostojakovicОценок пока нет
- AP Bio CH 14 Gene ExpressionДокумент24 страницыAP Bio CH 14 Gene ExpressionLuisa RamosОценок пока нет
- Virtual Western Blotting LabДокумент5 страницVirtual Western Blotting Labapi-522847737Оценок пока нет
- Genetic Code: Maria Septiana Parmonang AroeanДокумент15 страницGenetic Code: Maria Septiana Parmonang AroeanGe'sh Na'shy TyneОценок пока нет
- Gateway PaperДокумент3 страницыGateway PaperDivya DharshiniОценок пока нет
- Protein Synthesis - WorksheetДокумент7 страницProtein Synthesis - WorksheetMaisha IslamОценок пока нет
- Heterologous Expression of Cellulase Genes in Natural Saccharomyces Cerevisiae StrainsДокумент14 страницHeterologous Expression of Cellulase Genes in Natural Saccharomyces Cerevisiae StrainsJessi BoschОценок пока нет
- Basic PathologyДокумент35 страницBasic Pathologyshinichi kudoОценок пока нет
- (Fungal Biology) Monika Schmoll, Christoph Dattenböck (Eds.) - Gene Expression Systems in Fungi - Advancements and Applications (2016, Springer International Publishing)Документ505 страниц(Fungal Biology) Monika Schmoll, Christoph Dattenböck (Eds.) - Gene Expression Systems in Fungi - Advancements and Applications (2016, Springer International Publishing)Bodhi DharmaОценок пока нет
- Introduction To Bioinformatics: Course 341 Department of Computing Imperial College, London Moustafa GhanemДокумент42 страницыIntroduction To Bioinformatics: Course 341 Department of Computing Imperial College, London Moustafa GhanemCSiti HanifahОценок пока нет
- Machine Learning For Designing Next-Generation mRNA TherapeuticsДокумент11 страницMachine Learning For Designing Next-Generation mRNA TherapeuticsJohnОценок пока нет
- Cardiovascular Research Convergence 2022Документ16 страницCardiovascular Research Convergence 2022Aditya SutarОценок пока нет
- Ribosom Kloroplas EMBJ 36 475Документ12 страницRibosom Kloroplas EMBJ 36 475NovyKedungWilutОценок пока нет
- Use of Bacteriophage T7 RNA Polymerase To Direct SelectiveДокумент18 страницUse of Bacteriophage T7 RNA Polymerase To Direct Selectiveoplat147Оценок пока нет
- Lecture9 Genetic Code & Protein SynthesisДокумент64 страницыLecture9 Genetic Code & Protein SynthesisConstance WongОценок пока нет
- Aberrant Astrocyte Protein Secretion Contributes To Altered Neuronal Development in Multiple Models of Neurodevelopmental DisordersДокумент36 страницAberrant Astrocyte Protein Secretion Contributes To Altered Neuronal Development in Multiple Models of Neurodevelopmental DisordersLeon PalomeraОценок пока нет
- Important Questions of TranscriptionДокумент3 страницыImportant Questions of TranscriptionAna FerreiraОценок пока нет
- Lesson Plan Protein SynthesisДокумент2 страницыLesson Plan Protein SynthesisQueencess Ara Torres100% (1)
- Protein Synthesis Race WorksheetДокумент2 страницыProtein Synthesis Race WorksheetHugo FloresОценок пока нет
- R/aliens: Discord PoliciesДокумент34 страницыR/aliens: Discord PoliciesRude GeorgeОценок пока нет
- Statement of Purpose (Stanford)Документ2 страницыStatement of Purpose (Stanford)Emily CribasОценок пока нет
- 00 Unit-1-Slides - HandoutsДокумент10 страниц00 Unit-1-Slides - HandoutsShy OnОценок пока нет