Вам также может понравиться
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (345)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
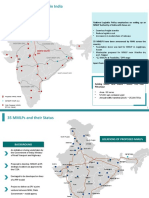
- Development of Mmlps in India: Western DFC Eastern DFCДокумент2 страницыDevelopment of Mmlps in India: Western DFC Eastern DFCsdfg100% (2)
- 1084662-Guia de Ivestigacion 2015 2016Документ108 страниц1084662-Guia de Ivestigacion 2015 2016Leochemical ChemicalОценок пока нет
- A Paleolimnological Perspective On Industrial-Era Metal Pollution in The Central Andes, PeruДокумент11 страницA Paleolimnological Perspective On Industrial-Era Metal Pollution in The Central Andes, PeruLeochemical ChemicalОценок пока нет
- What Really Sank The Titanic?Документ1 страницаWhat Really Sank The Titanic?Leochemical ChemicalОценок пока нет
- Journal Hydrometallurgy Solvent ExtractionДокумент13 страницJournal Hydrometallurgy Solvent ExtractionLeochemical ChemicalОценок пока нет
- Vocabulary Levels Tests Versions 1 2Документ12 страницVocabulary Levels Tests Versions 1 2Rangsiya PjewОценок пока нет
- Rociadores - FT - GFS-100B - GL SeriesДокумент2 страницыRociadores - FT - GFS-100B - GL SeriesJimmy FernándezОценок пока нет
- Understanding Terrorism and Political Violence PDFДокумент304 страницыUnderstanding Terrorism and Political Violence PDFmihaela buzatuОценок пока нет
- Background of The Study: Than IdealДокумент3 страницыBackground of The Study: Than IdealClint CamilonОценок пока нет
- TGA Interpretation of Data, Sources of ErrorДокумент28 страницTGA Interpretation of Data, Sources of ErrorUsman GhaniОценок пока нет
- Getting Good Grades in School Is What Kids Are Supposed To Be Doing.Документ6 страницGetting Good Grades in School Is What Kids Are Supposed To Be Doing.The QUEENОценок пока нет
- Spring 2010 MidTerm OPKST CS101 Bc100200572Документ6 страницSpring 2010 MidTerm OPKST CS101 Bc100200572cs619finalproject.comОценок пока нет
- A Practical Guide To HL7 Interface DevelopmentДокумент5 страницA Practical Guide To HL7 Interface DevelopmentmjohnstnОценок пока нет
- Financial Accounting Report (Partnership - Group 2)Документ20 страницFinancial Accounting Report (Partnership - Group 2)syednaim0300Оценок пока нет
- TA-Modulator EN LowДокумент16 страницTA-Modulator EN Lowkap4enijОценок пока нет
- Hydrogen Sulfide and Mercaptan Sulfur in Liquid Hydrocarbons by Potentiometric TitrationДокумент8 страницHydrogen Sulfide and Mercaptan Sulfur in Liquid Hydrocarbons by Potentiometric TitrationINOPETRO DO BRASILОценок пока нет
- Barangay Labangon Shelter Plan: Group 6 Blackjacks Ar 3134 HousingДокумент21 страницаBarangay Labangon Shelter Plan: Group 6 Blackjacks Ar 3134 HousingGicelle SenoОценок пока нет
- EagleBurgmann H7N ENДокумент5 страницEagleBurgmann H7N ENlamtony2013Оценок пока нет
- Lecture Note On Photovoltaic CellДокумент1 страницаLecture Note On Photovoltaic CellHaseeb NawazОценок пока нет
- Autodesk 3ds Max SkillsДокумент18 страницAutodesk 3ds Max SkillsJuan UrdanetaОценок пока нет
- Manual of Armacad v9 PDFДокумент102 страницыManual of Armacad v9 PDFCristiana FelicianoОценок пока нет
- BFC+43103. 1213 SEM1pdfДокумент19 страницBFC+43103. 1213 SEM1pdfAdibah Azimat100% (1)
- Usama Lab 6Документ8 страницUsama Lab 6M mubeen riazОценок пока нет
- Audio Level Meter - ProjectДокумент4 страницыAudio Level Meter - ProjectMircea PanzariuОценок пока нет
- Industrial ReactorsДокумент10 страницIndustrial ReactorssarahОценок пока нет
- Language Focus. Past Simple or Past ContinuousДокумент3 страницыLanguage Focus. Past Simple or Past ContinuoustotydnrОценок пока нет
- University of Central Punjab: Object Oriented ProgrammingДокумент3 страницыUniversity of Central Punjab: Object Oriented ProgrammingChoudhary MuneebОценок пока нет
- Vr31a OmДокумент5 страницVr31a OmrudydanielleОценок пока нет
- Optimal Control Development System For ElectricalДокумент7 страницOptimal Control Development System For ElectricalCRISTIAN CAMILO MORALES SOLISОценок пока нет
- Ai R16 - Unit-6Документ36 страницAi R16 - Unit-6RakeshОценок пока нет
- BSBPMG632 Manage Program RiskДокумент221 страницаBSBPMG632 Manage Program RiskgurpreetОценок пока нет
- Bài Tập Bổ Trợ Nâng Cao Tiếng Anh 7-8-9 Chương Trình Mới (1) -Trang-292-433Документ142 страницыBài Tập Bổ Trợ Nâng Cao Tiếng Anh 7-8-9 Chương Trình Mới (1) -Trang-292-433Nguyễn Lâm ThươngОценок пока нет
- Extenso MeterДокумент8 страницExtenso MeterVijayanandh Raja100% (1)