Вам также может понравиться
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (345)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- kx0804 PDFДокумент519 страницkx0804 PDFstefan corjuc100% (7)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- RFP Iecc KochiДокумент73 страницыRFP Iecc KochiCUBE ProjectsОценок пока нет
- Service Station Manual Vespa LX 125 - 150 4t Euro 3Документ241 страницаService Station Manual Vespa LX 125 - 150 4t Euro 3Adèle Standard100% (1)
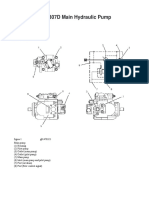
- Cat 307D Main Pump OperationДокумент3 страницыCat 307D Main Pump Operationkahandawala100% (1)
- ATPL Notes - ElectricsДокумент23 страницыATPL Notes - ElectricsMoslem Grimaldi100% (3)
- Deck Cranes PDFДокумент7 страницDeck Cranes PDFIndra Ranu KusumaОценок пока нет
- d20 ThundarrДокумент27 страницd20 ThundarrAlebrije1100% (1)
- High Pressure Drilling Hoses: Fleximak Industrial CatalogueДокумент5 страницHigh Pressure Drilling Hoses: Fleximak Industrial CatalogueitangОценок пока нет
- Design of Falling Film Evaporators U09CH152-U09CH157Документ7 страницDesign of Falling Film Evaporators U09CH152-U09CH157Kailasham RamalingamОценок пока нет
- Lenard Jones PotentialДокумент3 страницыLenard Jones PotentialKailasham RamalingamОценок пока нет
- Monte CarloДокумент21 страницаMonte CarloKailasham RamalingamОценок пока нет
- Molecular DynamicsДокумент26 страницMolecular DynamicsKailasham RamalingamОценок пока нет
- Lattice DynamicsДокумент16 страницLattice DynamicsKailasham RamalingamОценок пока нет
- Covalent BondingДокумент7 страницCovalent BondingKailasham RamalingamОценок пока нет
- Pair PotentialsДокумент10 страницPair PotentialsKailasham RamalingamОценок пока нет
- Nonlinear PDE SolverДокумент33 страницыNonlinear PDE SolverKailasham RamalingamОценок пока нет
- ODE Solver in MATLABДокумент15 страницODE Solver in MATLABKailasham RamalingamОценок пока нет
- Manual Grindex - Bravo400.Документ26 страницManual Grindex - Bravo400.Roque LlamoccaОценок пока нет
- Manual Gefran 600-1Документ4 страницыManual Gefran 600-1Eder AlexandreОценок пока нет
- Quantum Phase Transitions: Alexander DanielsДокумент12 страницQuantum Phase Transitions: Alexander Danielsapi-288833495Оценок пока нет
- Maintenance Issues of Photovoltaic System: Presented byДокумент10 страницMaintenance Issues of Photovoltaic System: Presented byjunaid bashirОценок пока нет
- Product Catalogue 2017: AquathermДокумент183 страницыProduct Catalogue 2017: AquathermZeeshanОценок пока нет
- Isv5 0-Web PDFДокумент2 страницыIsv5 0-Web PDFperulapiaОценок пока нет
- 6.automobile Engineering Lab - IДокумент7 страниц6.automobile Engineering Lab - IAmrithОценок пока нет
- Module3 PDFДокумент174 страницыModule3 PDFAhallya JaladeepОценок пока нет
- 1GR-FE ENGINE CONTROL: SFI SYSTEM: P0340, P0342, P0343, P0345, P0347, P0348: Camshaft Position Sensor Circuit MalfunctionДокумент12 страниц1GR-FE ENGINE CONTROL: SFI SYSTEM: P0340, P0342, P0343, P0345, P0347, P0348: Camshaft Position Sensor Circuit MalfunctionwilliamОценок пока нет
- KJLC Ed09 Sec09 Web200910Документ54 страницыKJLC Ed09 Sec09 Web200910NickMoloОценок пока нет
- Approximation Methods: Physics 130B, UCSD Fall 2009Документ90 страницApproximation Methods: Physics 130B, UCSD Fall 2009Luz PeñaОценок пока нет
- Design and Simulation of A Sine Wave Inverter With PID Control For Nonlinear Load ApplicationsДокумент12 страницDesign and Simulation of A Sine Wave Inverter With PID Control For Nonlinear Load ApplicationsReno ReoОценок пока нет
- Geometric and Physical Optics: InterferenceДокумент35 страницGeometric and Physical Optics: InterferenceHelenОценок пока нет
- UT Dallas Syllabus For Phys3341.001.11f Taught by Mark Lee (mxl101000)Документ5 страницUT Dallas Syllabus For Phys3341.001.11f Taught by Mark Lee (mxl101000)UT Dallas Provost's Technology GroupОценок пока нет
- ELL-332 Electric Drives Lecture 3: Different Types of Load TorquesДокумент25 страницELL-332 Electric Drives Lecture 3: Different Types of Load TorquesNikhil KumarОценок пока нет
- 8015-0151-ENTR-41-411-EL-MS-41205 - A0 Method Statement For Cathodic Protection PDFДокумент9 страниц8015-0151-ENTR-41-411-EL-MS-41205 - A0 Method Statement For Cathodic Protection PDFCripoОценок пока нет
- CMG StarsДокумент2 страницыCMG StarsSedighi100% (1)
- Presentation Material BalanceДокумент101 страницаPresentation Material BalanceSharizada KanapiyevaОценок пока нет
- Unit 1 Solar EnergyДокумент176 страницUnit 1 Solar EnergyAkhil Dayalu100% (1)
- TM 9 4935 601 14 3&PДокумент137 страницTM 9 4935 601 14 3&Pkhaerul jannahОценок пока нет
- Fayat Activity Report 2020 - en - Page by Page - Low Resolution - 0Документ44 страницыFayat Activity Report 2020 - en - Page by Page - Low Resolution - 0arlyОценок пока нет
- Karino Taani 2019Документ13 страницKarino Taani 2019AliОценок пока нет