Вам также может понравиться
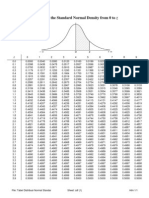
- Tabel Distribusi Normal BakuДокумент1 страницаTabel Distribusi Normal BakuAnonymous KUuLddnO9867% (6)
- Curve List Dan ObservasiДокумент5 страницCurve List Dan Observasieriska yunita sariОценок пока нет
- Cell Biology and CancerДокумент17 страницCell Biology and Cancersatya_chagantiОценок пока нет
- Family PlanningДокумент12 страницFamily Planningeriska yunita sariОценок пока нет
- Pertumbuhan Mikroba1Документ26 страницPertumbuhan Mikroba1eriska yunita sariОценок пока нет
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- Gmo - PPT BiotechДокумент16 страницGmo - PPT BiotechGerwyn Gervacio CОценок пока нет
- Student Manual pGLO Transformation: Lesson 1 Introduction To TransformationДокумент26 страницStudent Manual pGLO Transformation: Lesson 1 Introduction To TransformationanamqadirОценок пока нет
- Chapter 3: Heredity and VariationДокумент4 страницыChapter 3: Heredity and VariationADY2022Оценок пока нет
- Ethical Issues in Genetic Engineering PDFДокумент2 страницыEthical Issues in Genetic Engineering PDFAhmad0% (1)
- Genetics Practice ProblemsДокумент3 страницыGenetics Practice ProblemsZeyad MoustafaОценок пока нет
- GEN3051 Lecture 1: Human Genes and Human Genetic DisordersДокумент5 страницGEN3051 Lecture 1: Human Genes and Human Genetic DisordersAlessander Leyendecker JuniorОценок пока нет
- Paula-Navas 13451 - DNA Profiling GizmoДокумент7 страницPaula-Navas 13451 - DNA Profiling Gizmopaula-navas 13451Оценок пока нет
- Molecular Biology IntroductionДокумент8 страницMolecular Biology IntroductionDANIELLA LOMA CAPONPONОценок пока нет
- Chromatin WebДокумент7 страницChromatin Webyerram nikithaОценок пока нет
- Life Sciences P1 Nov 2012 Version 2 Memo EngДокумент10 страницLife Sciences P1 Nov 2012 Version 2 Memo EngedwardnephОценок пока нет
- Mutant Germplasm Characterization Using Molecular Markers: A ManualДокумент86 страницMutant Germplasm Characterization Using Molecular Markers: A ManualBlaxez YTОценок пока нет
- What Is RibotypingДокумент8 страницWhat Is RibotypingRobotrix0% (1)
- Extra Nuclear Inheritance: Transmission of Non Nuclear Genes. Mitochondria and ChloroplastsДокумент20 страницExtra Nuclear Inheritance: Transmission of Non Nuclear Genes. Mitochondria and ChloroplastsAvick DasОценок пока нет
- Buttross Susan Understanding ADHD PDFДокумент141 страницаButtross Susan Understanding ADHD PDFRuth Gasparini50% (2)
- 1st Week History Cytology LectureДокумент34 страницы1st Week History Cytology Lectureterryortiz825Оценок пока нет
- Physical Mapping of DNA: Balcera, Katrina Marie Hepiga, Adah MaeДокумент15 страницPhysical Mapping of DNA: Balcera, Katrina Marie Hepiga, Adah MaeKarshey Alagad ObutОценок пока нет
- Genetics Problems - Grade 11: Monohybrid CrossesДокумент4 страницыGenetics Problems - Grade 11: Monohybrid CrossessiennaОценок пока нет
- Phylogeny and Classification, Student Learning: Name: - Period: - DateДокумент3 страницыPhylogeny and Classification, Student Learning: Name: - Period: - DateL ChanОценок пока нет
- BS en 12687 - 1998Документ12 страницBS en 12687 - 1998Emanuele MastrangeloОценок пока нет
- Genome, Transcriptome and Proteome PDFДокумент17 страницGenome, Transcriptome and Proteome PDFAdn CodeОценок пока нет
- Journal Homepage: - : IntroductionДокумент8 страницJournal Homepage: - : IntroductionIJAR JOURNALОценок пока нет
- Cell Division Mitosis For Guided Notes Powerpoint 11 3 17Документ20 страницCell Division Mitosis For Guided Notes Powerpoint 11 3 17api-262586446100% (1)
- 1st Periodical Test Grade 9Документ7 страниц1st Periodical Test Grade 9Jay Ronnie PranadaОценок пока нет
- Blotting TechniquesДокумент36 страницBlotting TechniquesRajanathan96% (46)
- S6 Biology SB August 2018Документ421 страницаS6 Biology SB August 2018UMUFASHA Ange Justifiee100% (1)
- Hales - A Primer On The Drosophila Model SystemДокумент28 страницHales - A Primer On The Drosophila Model SystemCramblerОценок пока нет
- Next Gen Breeding Strategies For Climate Ready CropsДокумент27 страницNext Gen Breeding Strategies For Climate Ready Cropshema3rОценок пока нет
- High School Biology Teacher's Guide PDFДокумент562 страницыHigh School Biology Teacher's Guide PDFnino jamero100% (2)
- Pemeriksaan Genomik Untuk Preiksi Kanker (MF 08092019) PrintДокумент73 страницыPemeriksaan Genomik Untuk Preiksi Kanker (MF 08092019) Printnurrofiqoh kurniaОценок пока нет
- Biology - Fly Lab ResultsДокумент17 страницBiology - Fly Lab ResultslanichungОценок пока нет