Вам также может понравиться
- ALTE CEFR Speaking Grid Tests2014Документ11 страницALTE CEFR Speaking Grid Tests2014higginscribdОценок пока нет
- ELP Language Biography Plurilingual Profile enДокумент5 страницELP Language Biography Plurilingual Profile enhigginscribdОценок пока нет
- Abstracts For The 27th Annual Scientific Meeting of The Society For Immunotherapy of Cancer (SITC) PDFДокумент71 страницаAbstracts For The 27th Annual Scientific Meeting of The Society For Immunotherapy of Cancer (SITC) PDFhigginscribdОценок пока нет
- ELP What Is A Portfolio ENДокумент1 страницаELP What Is A Portfolio ENhigginscribdОценок пока нет
- ELP Language Biography Generic Checklists enДокумент18 страницELP Language Biography Generic Checklists enhigginscribdОценок пока нет
- Catalog Sep 2014Документ14 страницCatalog Sep 2014higginscribdОценок пока нет
- ELP Language Biography Checklists For Young Learners enДокумент8 страницELP Language Biography Checklists For Young Learners enhigginscribdОценок пока нет
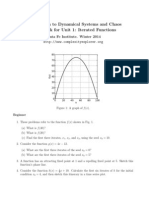
- Introduction To Dynamical Systems and Chaos Homework For Unit 1: Iterated FunctionsДокумент2 страницыIntroduction To Dynamical Systems and Chaos Homework For Unit 1: Iterated FunctionshigginscribdОценок пока нет
- Luraghi Narrog 4 Luraghi-LibreДокумент76 страницLuraghi Narrog 4 Luraghi-LibrehigginscribdОценок пока нет
- Praepositiones LatinaeДокумент1 страницаPraepositiones LatinaeMagister Optimus MaximusОценок пока нет
- 02.04.quiz 1 SolutionsДокумент1 страница02.04.quiz 1 SolutionshigginscribdОценок пока нет
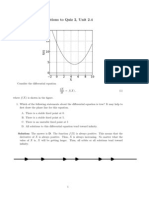
- Solutions To Quiz 2, Unit 2.4Документ1 страницаSolutions To Quiz 2, Unit 2.4higginscribdОценок пока нет
- Solutions To Quiz 1, Unit 2.2Документ1 страницаSolutions To Quiz 1, Unit 2.2higginscribdОценок пока нет
- Differential Equations: Some Notes On Terminology: DX DT F (X)Документ4 страницыDifferential Equations: Some Notes On Terminology: DX DT F (X)higginscribdОценок пока нет
- 02.05AlgorithmicSolutions Summary 2Документ3 страницы02.05AlgorithmicSolutions Summary 2higginscribdОценок пока нет
- What Are Complex Systems?Документ18 страницWhat Are Complex Systems?higginscribdОценок пока нет
- Self-Organization and Cooperation in Social SystemsДокумент29 страницSelf-Organization and Cooperation in Social SystemshigginscribdОценок пока нет
- Cellular Automata Are Idealized Models of Complex SystemsДокумент58 страницCellular Automata Are Idealized Models of Complex SystemshigginscribdОценок пока нет
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- RagragsakanДокумент6 страницRagragsakanRazel Hijastro86% (7)
- Learning Module 4 - BARTENDINGДокумент34 страницыLearning Module 4 - BARTENDINGivy mae flores67% (3)
- Modfied Indian Systems of MedicineДокумент8 страницModfied Indian Systems of MedicineNishamolKSОценок пока нет
- The Appropriate Biochemical Oxygen Demand Concentration For Designing Domestic Wastewater Treatment PlantДокумент8 страницThe Appropriate Biochemical Oxygen Demand Concentration For Designing Domestic Wastewater Treatment PlantabdulОценок пока нет
- Title Toolbox 1 AДокумент2 страницыTitle Toolbox 1 AGet LiveHelpОценок пока нет
- Openstack Deployment Ops Guide PDFДокумент197 страницOpenstack Deployment Ops Guide PDFBinank PatelОценок пока нет
- Maulana Azad Library, Aligarh Muslim UniversityДокумент38 страницMaulana Azad Library, Aligarh Muslim Universitybijesh babuОценок пока нет
- Division of Genetics ICAR-Indian Agricultural Research Institute, New Delhi - 110012Документ9 страницDivision of Genetics ICAR-Indian Agricultural Research Institute, New Delhi - 110012Shivam PateriyaОценок пока нет
- Jeevan Tara, Sansad Marg NEW DELHI-11001 Regonal Office (North Zone) E MailДокумент3 страницыJeevan Tara, Sansad Marg NEW DELHI-11001 Regonal Office (North Zone) E MailGourav SharmaОценок пока нет
- Resume For Singapore Spass Civil EngineerДокумент8 страницResume For Singapore Spass Civil EngineerArul SD100% (1)
- Campbell Smith - Harris032311Документ2 страницыCampbell Smith - Harris032311Heather X RhodesОценок пока нет
- Demand, Elasticity of Demand and Demand ForecastingДокумент16 страницDemand, Elasticity of Demand and Demand Forecastingankit thapliyal100% (1)
- STAS 111 - Information AgeДокумент20 страницSTAS 111 - Information AgeMayeee GayosoОценок пока нет
- Iit Delhi Project Scientist Project Associate Posts Advt Details Efa5f6Документ1 страницаIit Delhi Project Scientist Project Associate Posts Advt Details Efa5f6SadanandОценок пока нет
- China Email ListДокумент3 страницыChina Email ListRosie Brown40% (5)
- Peter Zumthor - Thinking Architecture PDFДокумент33 страницыPeter Zumthor - Thinking Architecture PDFDiana Sterian73% (11)
- Professional Education: St. Louis Review Center, IncДокумент10 страницProfessional Education: St. Louis Review Center, IncEarshad Shinichi IIIОценок пока нет
- Bcos 186Документ3 страницыBcos 186Shiv KumarОценок пока нет
- 10 Biological-HazardsДокумент31 страница10 Biological-HazardsjvОценок пока нет
- Motivation MBAДокумент31 страницаMotivation MBAAkshitaОценок пока нет
- PP Planning Workshop SAPДокумент36 страницPP Planning Workshop SAPDavid100% (1)
- A Crude Awakening Video NotesДокумент3 страницыA Crude Awakening Video NotesTai NguyenОценок пока нет
- Projects & Operations: IN: NE Power Systm ImprvmДокумент5 страницProjects & Operations: IN: NE Power Systm ImprvmGaurang PatelОценок пока нет
- Intro To Semiology Reading NotesДокумент6 страницIntro To Semiology Reading NotesRyan DrakeОценок пока нет
- The Magical Diaries of Ethel ArcherДокумент7 страницThe Magical Diaries of Ethel Archerleeghancock100% (1)
- III Job Order CostingДокумент66 страницIII Job Order CostingJoshuaGuerrero0% (1)
- Jobsheet PMДокумент49 страницJobsheet PMwindhy fitrianaОценок пока нет
- Sticker BookДокумент66 страницSticker BookIvan SutlovicОценок пока нет
- Class 11 Assignment 10 (Prac)Документ3 страницыClass 11 Assignment 10 (Prac)9crollno14bhewensagarsahuОценок пока нет
- Bearing Capacity of Closed and Open Ended Piles Installed in Loose Sand PDFДокумент22 страницыBearing Capacity of Closed and Open Ended Piles Installed in Loose Sand PDFAnonymous 8KOUFYqОценок пока нет