Вам также может понравиться
- Crayon ReportДокумент4 страницыCrayon ReportAiza CabolesОценок пока нет
- Delta To Wye and V.VДокумент4 страницыDelta To Wye and V.VAiza CabolesОценок пока нет
- Concepts and Models of Inorganic Chemistry DouglasДокумент9 страницConcepts and Models of Inorganic Chemistry DouglasKiran MandalОценок пока нет
- Powder Metallurgy Stainless Steels - Processing Microstructures, and PropertiesДокумент227 страницPowder Metallurgy Stainless Steels - Processing Microstructures, and PropertiesSamuel TesfayeОценок пока нет
- AluminiumДокумент3 страницыAluminiumKin Fatt Wong100% (1)
- FlotationДокумент39 страницFlotationMuhammad Zubair SharifОценок пока нет
- Synthesis of N-Chloroquinolines and N-Ethynylquinolines (nZ2, 4, 8) : Homo and Heterocoupling Reactions.Документ10 страницSynthesis of N-Chloroquinolines and N-Ethynylquinolines (nZ2, 4, 8) : Homo and Heterocoupling Reactions.kawtherahmedОценок пока нет
- Etard ReactionДокумент5 страницEtard Reactionp3pumОценок пока нет
- Asian J. Org. Chem. 2015, 4, 28 - 32 PDFДокумент5 страницAsian J. Org. Chem. 2015, 4, 28 - 32 PDFSulagna DasОценок пока нет
- A New Method For The Synthesis of Aliphatic Nitro Compounds1, 2Документ5 страницA New Method For The Synthesis of Aliphatic Nitro Compounds1, 2banjo01Оценок пока нет
- 1,5-Dipolar Cyclizations: 1. LntroducfionДокумент51 страница1,5-Dipolar Cyclizations: 1. LntroducfionRikta SahaОценок пока нет
- Nitrosation of Peptide BondДокумент7 страницNitrosation of Peptide BondEugenia CebanОценок пока нет
- Lewis Acid Promoted Reactions of N - (Chlorides. Ring-Size Effects in Competitive Intramolecular Acylation of Phenyl and Cyclopropyl SubstituentsДокумент3 страницыLewis Acid Promoted Reactions of N - (Chlorides. Ring-Size Effects in Competitive Intramolecular Acylation of Phenyl and Cyclopropyl SubstituentsthamtusieuquayОценок пока нет
- Transition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesОт EverandTransition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesОценок пока нет
- Thiophenol SynthesisДокумент6 страницThiophenol SynthesisNirav ShahОценок пока нет
- Expedient Access To Unsymmetrical Diarylindolylmethanes Through Palladium-Catalyzed Domino Electrophilic Cyclization Extended Conjugate Addition ApproachДокумент4 страницыExpedient Access To Unsymmetrical Diarylindolylmethanes Through Palladium-Catalyzed Domino Electrophilic Cyclization Extended Conjugate Addition ApproachMR.ZUHAIB MughalОценок пока нет
- Decompositions of Di-t-Alkyl Peroxides. I. Kinetics: Frederick F. EДокумент7 страницDecompositions of Di-t-Alkyl Peroxides. I. Kinetics: Frederick F. Emartinml_1191Оценок пока нет
- Synthesis of Levulinic Acid-Glycerol Ketal-Ester Oligomers and Structural Characterization Using NMR SpectrosДокумент7 страницSynthesis of Levulinic Acid-Glycerol Ketal-Ester Oligomers and Structural Characterization Using NMR SpectrosLucas de MeloОценок пока нет
- Synthesis and Evaluation of Some Variants of The Nefkens' ReagentДокумент3 страницыSynthesis and Evaluation of Some Variants of The Nefkens' Reagentlost6taОценок пока нет
- New Montmorillonite Silylpropylethylenediamine Palladium (II) Complex in Oxidation of Terminal OlefinsДокумент6 страницNew Montmorillonite Silylpropylethylenediamine Palladium (II) Complex in Oxidation of Terminal OlefinsChamula K MasОценок пока нет
- Chem. Eur. J. 2011, 17, 10208 - 10212Документ5 страницChem. Eur. J. 2011, 17, 10208 - 10212SBОценок пока нет
- 1 s2.0 S0926860X08000963 MainДокумент9 страниц1 s2.0 S0926860X08000963 Mainpetru apopeiОценок пока нет
- Kinetics of The Ring-Opening Polymerization Of, - Lactide Using Zinc (II) Octoate As CatalystДокумент9 страницKinetics of The Ring-Opening Polymerization Of, - Lactide Using Zinc (II) Octoate As CatalystGift GibbОценок пока нет
- Tetrahedron Letters: Shan Wang, Jian Wang, Rui Guo, Gao Wang, Shan-Yong Chen, Xiao-Qi YuДокумент4 страницыTetrahedron Letters: Shan Wang, Jian Wang, Rui Guo, Gao Wang, Shan-Yong Chen, Xiao-Qi Yumarkbronson2009Оценок пока нет
- The Role and Reactions of Nitroxyl Radicals in Hindered Piperidine Light StabilisationДокумент8 страницThe Role and Reactions of Nitroxyl Radicals in Hindered Piperidine Light StabilisationStacey DongОценок пока нет
- Paper: Microwave Methods For The Synthesis of Paddlewheel Diruthenium Compounds With N, N-Donor LigandsДокумент6 страницPaper: Microwave Methods For The Synthesis of Paddlewheel Diruthenium Compounds With N, N-Donor LigandshttmaiОценок пока нет
- P 19900000945Документ10 страницP 19900000945Yi-Yan TsaoОценок пока нет
- Kinetics of Iodination of Acetone Catalyzed by HCL and H2so4 A Colorimetric Investigation of Relative StrengthДокумент2 страницыKinetics of Iodination of Acetone Catalyzed by HCL and H2so4 A Colorimetric Investigation of Relative StrengthHansel VereitelnОценок пока нет
- PD CatДокумент7 страницPD CatKiss LeviОценок пока нет
- Review of Blocked and Deblocked IsocyanateДокумент8 страницReview of Blocked and Deblocked IsocyanateAdlyLubisОценок пока нет
- R.A. Heacock and M.E. Mahon - The Chemistry of The "Aminochromes" Part II: The Preparation, Paper Chromatography, and Spectroscopic Properties of Pure Adrenolutin The Infrared Spectrum of AdrenochromeДокумент5 страницR.A. Heacock and M.E. Mahon - The Chemistry of The "Aminochromes" Part II: The Preparation, Paper Chromatography, and Spectroscopic Properties of Pure Adrenolutin The Infrared Spectrum of AdrenochromeGummyColaОценок пока нет
- An Experiment For Undergraduate Advanced Inorganic Chemistry StudentsДокумент19 страницAn Experiment For Undergraduate Advanced Inorganic Chemistry StudentsKiki AimaОценок пока нет
- A Practical Synthesis of (-) - Kainic AcidДокумент11 страницA Practical Synthesis of (-) - Kainic AcidNikhil SarpateОценок пока нет
- Com 97 8051Документ5 страницCom 97 8051bruna_0410Оценок пока нет
- 1971 R K Kulkarni E G Moore A F Hegyeli - Fred Leonard (1971) - Biodegradable Poly (Lactic Acid) Polymers 5 (3) 169-181 1971Документ13 страниц1971 R K Kulkarni E G Moore A F Hegyeli - Fred Leonard (1971) - Biodegradable Poly (Lactic Acid) Polymers 5 (3) 169-181 1971Julio ArruaОценок пока нет
- Titanium FractionДокумент9 страницTitanium FractionAhmadОценок пока нет
- Tetrahedron Letters Volume Issue 2016 (Doi 10.1016/j.tetlet.2016.04.112) Nagarajan, Rajendran Jayashankaran, Jayadevan Emmanuvel, Lourd - Transition Metal-Free Steric Controlled One - Pot SynthesДокумент13 страницTetrahedron Letters Volume Issue 2016 (Doi 10.1016/j.tetlet.2016.04.112) Nagarajan, Rajendran Jayashankaran, Jayadevan Emmanuvel, Lourd - Transition Metal-Free Steric Controlled One - Pot SynthesJayash RulzОценок пока нет
- Mo JAEДокумент4 страницыMo JAEThanhThao TranОценок пока нет
- Angew. Chem. Int. Ed. 2011, 50, 6167 - 6170Документ4 страницыAngew. Chem. Int. Ed. 2011, 50, 6167 - 6170NoimurОценок пока нет
- Preparation of A Polysulfide: RubberДокумент1 страницаPreparation of A Polysulfide: RubberRicky EstepaОценок пока нет
- A Review of Organotin Compounds Chemistry and ApplДокумент9 страницA Review of Organotin Compounds Chemistry and ApplJuan C HernandezОценок пока нет
- Datta 2012Документ4 страницыDatta 2012SakhaviTVОценок пока нет
- Aryl GrignardДокумент4 страницыAryl GrignardRoss LewinОценок пока нет
- Glyconanoparticles Activity 3Документ3 страницыGlyconanoparticles Activity 3ЛуизАпазаТ.Оценок пока нет
- Kim2002 - PolikondensasiДокумент6 страницKim2002 - PolikondensasiMathilda PasaribuОценок пока нет
- 檔案下載Документ6 страниц檔案下載Rosalina DjatmikaОценок пока нет
- The Room Temperature Polymerization of Propylene OxideДокумент5 страницThe Room Temperature Polymerization of Propylene OxidecesarmachucaОценок пока нет
- Synthesis, Structure and Chemical Transformations of 4-AminobenzaldehydeДокумент5 страницSynthesis, Structure and Chemical Transformations of 4-AminobenzaldehydeBrem BalazsОценок пока нет
- FeCl3-Catalyzed Synthesis of 2-Methyl-4-Substituted-1,2,3,4-Tetrahydroquinoline DerivativesДокумент4 страницыFeCl3-Catalyzed Synthesis of 2-Methyl-4-Substituted-1,2,3,4-Tetrahydroquinoline DerivativesRajesh TammanaОценок пока нет
- Asian J. Org. Chem. 2021,10, 2152-2156Документ5 страницAsian J. Org. Chem. 2021,10, 2152-2156NoimurОценок пока нет
- Isolation, Biological Activities and Synthesis of Indoloquinoline Alkaloids: Cryptole-Pine, Isocryptolepine and NeocryptolepineДокумент22 страницыIsolation, Biological Activities and Synthesis of Indoloquinoline Alkaloids: Cryptole-Pine, Isocryptolepine and NeocryptolepineDayse_sbОценок пока нет
- Tetrahedron 64 (2008) 219e233 - RosyДокумент15 страницTetrahedron 64 (2008) 219e233 - RosyRamdas BorhadeОценок пока нет
- Exp't 13: Phase-Transfer-Catalyzed Alkylation of Diethyl MalonateДокумент5 страницExp't 13: Phase-Transfer-Catalyzed Alkylation of Diethyl MalonatelovehopeОценок пока нет
- ReductionДокумент2 страницыReductionCatenaneОценок пока нет
- tmp2AEF TMPДокумент6 страницtmp2AEF TMPFrontiersОценок пока нет
- Synthesis, Characterization and Application of O, O-Diethyl Acrylamide PhosphateДокумент5 страницSynthesis, Characterization and Application of O, O-Diethyl Acrylamide Phosphateandi tiaОценок пока нет
- Chiral Sulphonated Phosphines. Part VII. Catalytic Transfer-Hydrogenation of Unsaturated Substrates With Formates in The Presence of Water Soluble Complexes of RhodaДокумент4 страницыChiral Sulphonated Phosphines. Part VII. Catalytic Transfer-Hydrogenation of Unsaturated Substrates With Formates in The Presence of Water Soluble Complexes of RhodappopgodОценок пока нет
- MR14 5 0510Документ7 страницMR14 5 0510rusheekesh3497Оценок пока нет
- (Grupo 3) Wang-2021-Ligandcontrolled-tunable-coppercataДокумент8 страниц(Grupo 3) Wang-2021-Ligandcontrolled-tunable-coppercataVANESSA RODRIGUEZ CEBALLOSОценок пока нет
- Strategies Toward The Design of Energetic Ionic Liquids: Nitro-And Nitrile-Substituted N, N - Dialkylimidazolium SaltswДокумент10 страницStrategies Toward The Design of Energetic Ionic Liquids: Nitro-And Nitrile-Substituted N, N - Dialkylimidazolium SaltswZhan FangОценок пока нет
- 12 - Xu - JMolCatA - Dihydrochalcones Via The AlCl3 CynamoylДокумент9 страниц12 - Xu - JMolCatA - Dihydrochalcones Via The AlCl3 Cynamoyllenggah purwandariОценок пока нет
- Standard Molar Enthalpies of Formation of Crystalline Stereoisomers of Aldono-1,4-LactonesДокумент6 страницStandard Molar Enthalpies of Formation of Crystalline Stereoisomers of Aldono-1,4-LactonesManuОценок пока нет
- Luminescence Probe Studies of Nafion PolyelectrolytesДокумент5 страницLuminescence Probe Studies of Nafion PolyelectrolytesLuis AlvarezОценок пока нет
- Binol On SilicaДокумент8 страницBinol On SilicaMinal ButalaОценок пока нет
- Sodium Nitrite Catalyzed Aerobic Oxidative Deoximation Under Mild ConditionsДокумент4 страницыSodium Nitrite Catalyzed Aerobic Oxidative Deoximation Under Mild ConditionsCláudio SerafimОценок пока нет
- Transition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesОт EverandTransition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesОценок пока нет
- Sun Life ProspectusДокумент39 страницSun Life ProspectusAiza CabolesОценок пока нет
- Critique PaperДокумент1 страницаCritique PaperAiza CabolesОценок пока нет
- MSDS Polyamine - Bluwat ChemicalsДокумент4 страницыMSDS Polyamine - Bluwat ChemicalsAiza CabolesОценок пока нет
- Base ConversionДокумент2 страницыBase ConversionAiza CabolesОценок пока нет
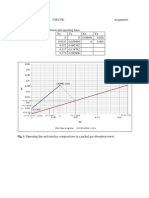
- Table 1. Data For The Equilibrium and Operating Lines.: Caboles, Ellaiza Marie G. Che153E Assignment 2011-45827Документ1 страницаTable 1. Data For The Equilibrium and Operating Lines.: Caboles, Ellaiza Marie G. Che153E Assignment 2011-45827Aiza CabolesОценок пока нет
- DR Perla D Santos OcampoДокумент2 страницыDR Perla D Santos OcampoAiza CabolesОценок пока нет
- Sample Calculations For Surface TensionДокумент6 страницSample Calculations For Surface TensionAiza CabolesОценок пока нет
- How To Calculate TorqueДокумент6 страницHow To Calculate TorqueAiza Caboles100% (1)
- Table 1. Values Computed For The Multicomponent Absorption DiagramДокумент1 страницаTable 1. Values Computed For The Multicomponent Absorption DiagramAiza CabolesОценок пока нет
- UOP ParaxyleneДокумент2 страницыUOP ParaxyleneAiza CabolesОценок пока нет
- ChE 192 Handout 6a (Storage Tank)Документ4 страницыChE 192 Handout 6a (Storage Tank)Aiza CabolesОценок пока нет
- Oxygen Bomb CalorimeterДокумент5 страницOxygen Bomb CalorimeterAiza CabolesОценок пока нет
- Experimental Determination of The Joule Thomson Coefficient of Nitrogen Carbon Dioxide and OxygenДокумент5 страницExperimental Determination of The Joule Thomson Coefficient of Nitrogen Carbon Dioxide and OxygenAiza CabolesОценок пока нет
- Eubp Factsfigures Bioplastics 2013Документ6 страницEubp Factsfigures Bioplastics 2013Aiza CabolesОценок пока нет
- Che 154 Lecture 3b Screening EquipmentДокумент34 страницыChe 154 Lecture 3b Screening EquipmentAiza CabolesОценок пока нет
- Hyouka V 1 (You Cant Escape - The Niece of Time)Документ159 страницHyouka V 1 (You Cant Escape - The Niece of Time)RinokukunОценок пока нет
- Fiberglass Composite MaterialsДокумент40 страницFiberglass Composite MaterialsIsaacColemanОценок пока нет
- Pickling Tubing StringsДокумент3 страницыPickling Tubing StringsHuong Viet100% (1)
- Kahoot!Документ12 страницKahoot!Ashiya GrierОценок пока нет
- Kitchen Chemistry Solubility Melting Point and ConductivityДокумент7 страницKitchen Chemistry Solubility Melting Point and ConductivityMss FaixaОценок пока нет
- 1-7 Manufacturing BasicsДокумент29 страниц1-7 Manufacturing Basicselektron2010Оценок пока нет
- Cambridge Checkpoint Science Skills Builder 8 AnswersДокумент17 страницCambridge Checkpoint Science Skills Builder 8 AnswersJaeikLee100% (3)
- Chemistry/Polymer Science: Category GENДокумент22 страницыChemistry/Polymer Science: Category GENShweta RamtekeОценок пока нет
- Equivalents of Carbon Steel QualitiesДокумент10 страницEquivalents of Carbon Steel Qualitiessentilmech07Оценок пока нет
- Practical Aspects of Production Bright Annealing of Stainless SteelДокумент2 страницыPractical Aspects of Production Bright Annealing of Stainless SteelAgniva DuttaОценок пока нет
- PIONIER 2076P Data SheetДокумент1 страницаPIONIER 2076P Data SheetAlexis GaydaОценок пока нет
- Laboratory SafetyДокумент2 страницыLaboratory Safetyann wamburaОценок пока нет
- COREX Technology PDFДокумент43 страницыCOREX Technology PDFSheila Mae GardonОценок пока нет
- Submerged Arc Welding (Saw)Документ26 страницSubmerged Arc Welding (Saw)Kurt1905Оценок пока нет
- Separation of Organic Compounds Using Liquid-Liquid ExtractionДокумент8 страницSeparation of Organic Compounds Using Liquid-Liquid ExtractionShyam BhaktaОценок пока нет
- 2542 Molygen Sae 5w-50 - enДокумент2 страницы2542 Molygen Sae 5w-50 - enAmy NolanОценок пока нет
- Periodic Classification of ElementsДокумент64 страницыPeriodic Classification of ElementsHarshit SwayamОценок пока нет
- K R I T I L E N® Masterbatches: Additives Technical InformationДокумент10 страницK R I T I L E N® Masterbatches: Additives Technical InformationAnas AbdoОценок пока нет
- Bonding in Organic Compounds - Organic Synthesis Marks SchemeДокумент96 страницBonding in Organic Compounds - Organic Synthesis Marks SchemeRaiyan RahmanОценок пока нет
- Rawat2020 Article Phosphate-SolubilizingMicroorgДокумент21 страницаRawat2020 Article Phosphate-SolubilizingMicroorgJessica Rodriguez EscobarОценок пока нет
- FA 1 Science 8th, 9th and 10thДокумент4 страницыFA 1 Science 8th, 9th and 10thRohithОценок пока нет
- Lec - 6Документ9 страницLec - 6warekarОценок пока нет
- Lithium - Brochure - ApprovedДокумент1 страницаLithium - Brochure - ApprovedVivek Ranganathan100% (1)
- Refining of Stainless SteelsДокумент27 страницRefining of Stainless SteelsirajfarjiОценок пока нет
- Water Shut Off by Rel Perm Modifiers - Lessons From Several FieldsДокумент14 страницWater Shut Off by Rel Perm Modifiers - Lessons From Several FieldsCarlos SuárezОценок пока нет
- Synthesis of Porous Hierarchical Mgo and Its Superb Adsorption PropertiesДокумент8 страницSynthesis of Porous Hierarchical Mgo and Its Superb Adsorption PropertiesJuan Diego CarrilloОценок пока нет
- Biokerosene and Green Diesel From Macauba Oils Via Catalytic Deoxygenation Over PDCДокумент10 страницBiokerosene and Green Diesel From Macauba Oils Via Catalytic Deoxygenation Over PDCLaura RDОценок пока нет