Вам также может понравиться
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- Data Documentation Power PlantsДокумент104 страницыData Documentation Power PlantsBabic UgljesaОценок пока нет
- Robust Calculation and Parameter Estimation of The Hourly Price Forward Curve PDFДокумент7 страницRobust Calculation and Parameter Estimation of The Hourly Price Forward Curve PDFBabic UgljesaОценок пока нет
- Lecture 4nДокумент50 страницLecture 4nBabic UgljesaОценок пока нет
- Aigul Issakova - InternshipДокумент25 страницAigul Issakova - InternshipBabic UgljesaОценок пока нет
- The Control Techniques Drives and Controls Handbook 2nd EditionДокумент765 страницThe Control Techniques Drives and Controls Handbook 2nd EditionSamuel Okuwobi94% (16)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- Thermoelasticity - WikiversityДокумент7 страницThermoelasticity - Wikiversityrpd25796145Оценок пока нет
- Tsu m23 Practice Problems Reinforced Concrete DesignДокумент2 страницыTsu m23 Practice Problems Reinforced Concrete DesignMark Lester ValdozОценок пока нет
- 02 - UNIT-II - Part I PDFДокумент72 страницы02 - UNIT-II - Part I PDFsrajan gupta100% (1)
- Evaluation For VesselДокумент10 страницEvaluation For VesselEngineering ESIIОценок пока нет
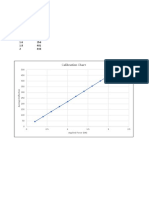
- Calibration Chart: Applied Force (KN)Документ4 страницыCalibration Chart: Applied Force (KN)Vishal ShrivastavaОценок пока нет
- Digimat 4.5.1 BrochureДокумент8 страницDigimat 4.5.1 BrochureEmily MillerОценок пока нет
- PTC Creo - Analyzing 3D StructuresДокумент20 страницPTC Creo - Analyzing 3D StructuresTommaso LeatiОценок пока нет
- Thick CylindersДокумент20 страницThick CylindersTayyab Zafar100% (1)
- PNC00004 PDFДокумент9 страницPNC00004 PDFRaymundo Maldonado AlvarezОценок пока нет
- Chitosan - ESTIC 2021Документ8 страницChitosan - ESTIC 2021faraz janatraОценок пока нет
- Finite Element Analysis and Calculation of HTC15J01 Petrochemical Industry Crane Pulley DesignДокумент6 страницFinite Element Analysis and Calculation of HTC15J01 Petrochemical Industry Crane Pulley DesignFrancisco Raro AlmuzaraОценок пока нет
- Modeling and Analysis of Wheel Rim Using AnsysДокумент4 страницыModeling and Analysis of Wheel Rim Using AnsysRahul GautamОценок пока нет
- Mech 1 AДокумент14 страницMech 1 AWin100% (1)
- Ansys TipsДокумент77 страницAnsys TipsFeby Philip AbrahamОценок пока нет
- MFT PPT (Autosaved)Документ432 страницыMFT PPT (Autosaved)Bibek DevОценок пока нет
- Entries M OДокумент46 страницEntries M OXiaohui ChenОценок пока нет
- Ansys TutorialДокумент25 страницAnsys TutorialRajeev RajeevОценок пока нет
- Som Question PaperДокумент6 страницSom Question PaperBalvinderОценок пока нет
- The Modeling of Chemical Reactors Chapter 7Документ17 страницThe Modeling of Chemical Reactors Chapter 7ManoakiОценок пока нет
- 247232333-Tubular-Joint-API-RP-2A-Design - Copie PDFДокумент49 страниц247232333-Tubular-Joint-API-RP-2A-Design - Copie PDFBayari ArОценок пока нет
- High Performance Machining For DielMold Manufacturing - R&D in ProgressДокумент22 страницыHigh Performance Machining For DielMold Manufacturing - R&D in ProgressJairo WilsonОценок пока нет
- Riveted JointsДокумент44 страницыRiveted Jointssharwan sharma67% (3)
- Shotcrete Support Load CalculationsДокумент6 страницShotcrete Support Load Calculationsanon_160157252Оценок пока нет
- Design of AbutmentДокумент39 страницDesign of AbutmentMukesh JangidОценок пока нет
- Cummins Pfleuger PaperДокумент13 страницCummins Pfleuger Papersachin_sawant1985Оценок пока нет
- Reinforcement Detailing of A CorbelДокумент13 страницReinforcement Detailing of A CorbelAchilles TroyОценок пока нет
- Mechanics of Elastic Solids - Lesson - TeachEngineering PDFДокумент7 страницMechanics of Elastic Solids - Lesson - TeachEngineering PDFIhsan BazirganОценок пока нет
- Strength IIДокумент172 страницыStrength IIክበር ተመስጌንОценок пока нет
- DocxДокумент9 страницDocxjamsheed sajidОценок пока нет
- Benchmarks PDFДокумент1 761 страницаBenchmarks PDFSyed Imtiaz Ali ShahОценок пока нет