Вам также может понравиться
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (894)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (265)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (119)
- Design of Predictive Magic Cards.Документ26 страницDesign of Predictive Magic Cards.aries25th3Оценок пока нет
- Mechanical Vibration NotesДокумент56 страницMechanical Vibration NotesYadanaОценок пока нет
- Chapter 3Документ7 страницChapter 3SULTAN SksaОценок пока нет
- DLL Mathematics-5 Q3 W5Документ7 страницDLL Mathematics-5 Q3 W5Charlota PelОценок пока нет
- 9789533073248UWBTechnologiesДокумент454 страницы9789533073248UWBTechnologiesConstantin PişteaОценок пока нет
- Risk, Return, and The Capital Asset Pricing ModelДокумент52 страницыRisk, Return, and The Capital Asset Pricing ModelFaryal ShahidОценок пока нет
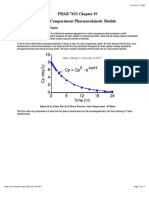
- PHAR 7633 Chapter 19 Multi-Compartment Pharmacokinetic ModelsДокумент23 страницыPHAR 7633 Chapter 19 Multi-Compartment Pharmacokinetic ModelsBandameedi RamuОценок пока нет
- The Cricket Winner Prediction With Applications of ML and Data AnalyticsДокумент18 страницThe Cricket Winner Prediction With Applications of ML and Data AnalyticsMuhammad SwalihОценок пока нет
- ATOA CAE Multiphysics and Multimaterial Design With COMSOL Webinar PДокумент31 страницаATOA CAE Multiphysics and Multimaterial Design With COMSOL Webinar PRaj C ThiagarajanОценок пока нет
- Optimal f ratio for inverter chainДокумент6 страницOptimal f ratio for inverter chainVIKAS RAOОценок пока нет
- Calculating Surface Integrals and Parameterizing SurfacesДокумент28 страницCalculating Surface Integrals and Parameterizing SurfacesKenn SharpeyesОценок пока нет
- G (X) F (X, Y) : Marginal Distributions Definition 5Документ13 страницG (X) F (X, Y) : Marginal Distributions Definition 5Kimondo KingОценок пока нет
- Rust Cheat SheetДокумент8 страницRust Cheat Sheetnewgmail accountОценок пока нет
- Divya MishraДокумент1 страницаDivya MishraKashish AwasthiОценок пока нет
- National University: Name/Section Rating Date Performed Date SubmittedДокумент4 страницыNational University: Name/Section Rating Date Performed Date SubmittedGail Domingo0% (1)
- Full Download Ebook Ebook PDF Mathematics in Action Algebraic Graphical and Trigonometric Problem Solving 5th Edition PDFДокумент42 страницыFull Download Ebook Ebook PDF Mathematics in Action Algebraic Graphical and Trigonometric Problem Solving 5th Edition PDFwillie.ortiz343100% (41)
- Javascript - Domain Fundamentals AssignmentsДокумент43 страницыJavascript - Domain Fundamentals AssignmentsSana Fathima SanaОценок пока нет
- Pump CavitationДокумент5 страницPump Cavitationjrri16Оценок пока нет
- Exam Circular - HYE - C1 To 12, 2023-24Документ26 страницExam Circular - HYE - C1 To 12, 2023-24Aman ShethОценок пока нет
- Brent Kung AdderДокумент60 страницBrent Kung AdderAnonymous gLVMeN2hОценок пока нет
- Bloomberg MIT Spring Tech TalkДокумент2 страницыBloomberg MIT Spring Tech TalkBita MoghaddamОценок пока нет
- Understandable Statistics Concepts and Methods 12th EditionДокумент61 страницаUnderstandable Statistics Concepts and Methods 12th Editionherbert.porter697100% (36)
- Ansys Coupled Structural Thermal AnalysisДокумент12 страницAnsys Coupled Structural Thermal AnalysisVaibhava Srivastava100% (1)
- Handout Diode EquationДокумент1 страницаHandout Diode Equationmanpreetsingh3458417Оценок пока нет
- Calculus SyllabusДокумент7 страницCalculus SyllabusRumarie de la CruzОценок пока нет
- BoualiS 3dДокумент8 страницBoualiS 3dGuilherme Francisco ComassettoОценок пока нет
- Assignment / Tugasan - Mathematics For ManagementДокумент7 страницAssignment / Tugasan - Mathematics For ManagementKetz NKОценок пока нет
- Uranian PlanetsДокумент12 страницUranian PlanetsPongwuthОценок пока нет
- Mock Test Paper 2013 (Answers)Документ84 страницыMock Test Paper 2013 (Answers)Varun GuptaОценок пока нет
- Graphs of Polynomial Functions: Digital LessonДокумент13 страницGraphs of Polynomial Functions: Digital LessonAna May BanielОценок пока нет