Вам также может понравиться
- Qué Es El Método CientíficoДокумент7 страницQué Es El Método CientíficoclajericoОценок пока нет
- Teorias Del Origen de La VidaДокумент7 страницTeorias Del Origen de La VidaclajericoОценок пока нет
- Qué Es ComputaciónДокумент8 страницQué Es ComputaciónclajericoОценок пока нет
- Sustantivos GentiliciosДокумент1 страницаSustantivos GentiliciosclajericoОценок пока нет
- SmartphoneДокумент4 страницыSmartphoneclajericoОценок пока нет
- Buscadores en InternetДокумент3 страницыBuscadores en InternetclajericoОценок пока нет
- Historia de Los Idiomas MayasДокумент4 страницыHistoria de Los Idiomas Mayasclajerico0% (1)
- Qué Es El AguaДокумент9 страницQué Es El AguaclajericoОценок пока нет
- Enfermedades Causadas Por VirusДокумент4 страницыEnfermedades Causadas Por Virusclajerico100% (1)
- La DurezaДокумент23 страницыLa DurezaclajericoОценок пока нет
- Frases VerbalesДокумент3 страницыFrases Verbalesclajerico100% (1)
- Mapa ConceptualДокумент2 страницыMapa ConceptualclajericoОценок пока нет
- Ciencias Relacionados Conla BiologiaДокумент10 страницCiencias Relacionados Conla BiologiaclajericoОценок пока нет
- Mejoramiento de Los Ingresos y La Economia FamiliarДокумент8 страницMejoramiento de Los Ingresos y La Economia FamiliarclajericoОценок пока нет
- Ciencias Relacionados Conla BiologiaДокумент10 страницCiencias Relacionados Conla BiologiaclajericoОценок пока нет
- PresidentesДокумент15 страницPresidentesclajericoОценок пока нет
- NEUROTRANSMISORДокумент21 страницаNEUROTRANSMISORclajericoОценок пока нет
- Períodos de La Historia EgipciaДокумент5 страницPeríodos de La Historia EgipciaclajericoОценок пока нет
- PlanificaciondocenteДокумент10 страницPlanificaciondocenteclajerico100% (3)
- Unidad FamiliarДокумент1 страницаUnidad FamiliarclajericoОценок пока нет
- Biografía de PresidentesДокумент7 страницBiografía de PresidentesclajericoОценок пока нет
- PresidentesДокумент15 страницPresidentesclajericoОценок пока нет
- Practica No. 6 Interaccion de FarmacosДокумент9 страницPractica No. 6 Interaccion de FarmacosMiguel PisteОценок пока нет
- 2 Farmacocin1Документ10 страниц2 Farmacocin1Liz VerónicaОценок пока нет
- Clase #5 FORMAS FARMACÉUTICAS Y VÍAS DE ADMINISTRACIÓNNДокумент30 страницClase #5 FORMAS FARMACÉUTICAS Y VÍAS DE ADMINISTRACIÓNNQF Carlos AbarcaОценок пока нет
- PK y PD en TIVA Curso Anual 2017Документ32 страницыPK y PD en TIVA Curso Anual 2017Aylud RaimeОценок пока нет
- Informe N°01 Practico de FarmacologíaДокумент24 страницыInforme N°01 Practico de FarmacologíaElida Delgado Vasquez100% (1)
- Bioequivalence Study of Tramadol by Intramuscular Administraitos in Healthy VolunterersДокумент5 страницBioequivalence Study of Tramadol by Intramuscular Administraitos in Healthy VolunterersJminsОценок пока нет
- Práctica 2. Muac - IVДокумент14 страницPráctica 2. Muac - IVJosseline SuarezОценок пока нет
- PROYECTO SEGUIR ACOPLANDO FinalДокумент55 страницPROYECTO SEGUIR ACOPLANDO FinalELIZAОценок пока нет
- Receta ImssДокумент1 страницаReceta Imssraul castillo gomezОценок пока нет
- Img 019Документ1 страницаImg 019Jeszii RojasОценок пока нет
- Taller Formas FarmacéuticasДокумент6 страницTaller Formas FarmacéuticasSebastian Rivera Alzate100% (1)
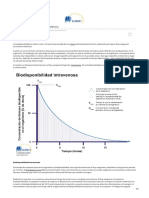
- BiodisponibilidadДокумент4 страницыBiodisponibilidadMargot Mendoza SalasОценок пока нет
- Farmacocinetica ExposicionДокумент30 страницFarmacocinetica Exposicioncarlos miguel mendoza mallmaОценок пока нет
- Administracion de Medicamentos Fraccionados PDFДокумент14 страницAdministracion de Medicamentos Fraccionados PDFAntonio TincopaОценок пока нет
- Forma FarmacéuticaДокумент2 страницыForma FarmacéuticaJorge ChaparroОценок пока нет
- Listado General de Medicamentos-201-246Документ60 страницListado General de Medicamentos-201-246pradoОценок пока нет
- Presentacion Corporativa InternacionalДокумент40 страницPresentacion Corporativa InternacionalFernando CendalesОценок пока нет
- Farmacocinetica y FarmacodinamiaДокумент77 страницFarmacocinetica y FarmacodinamiaAna Cítrico75% (4)
- Vías de Administración y Formas FarmacéuticasДокумент23 страницыVías de Administración y Formas FarmacéuticasKatty Bonilla OrtizОценок пока нет
- Farmacología clínica: farmacocinética y farmacodinamiaДокумент109 страницFarmacología clínica: farmacocinética y farmacodinamiaavengersrm 2019Оценок пока нет
- Acta 51 Guia BD y Be 1997Документ40 страницActa 51 Guia BD y Be 1997emunoz100Оценок пока нет
- Administración de MedicamentosДокумент9 страницAdministración de MedicamentosJose Fabian Cobeña ZambranoОценок пока нет
- Ladme y Vias de AdministracionДокумент42 страницыLadme y Vias de AdministracionErick KnoxОценок пока нет
- Tema 7 Dispensacion de Productos FarmaceuticosДокумент16 страницTema 7 Dispensacion de Productos FarmaceuticosRaquel Gonzalez RodriguezОценок пока нет
- Nom 257Документ10 страницNom 257Omar Michel GonzalezОценок пока нет
- Códigos InactivosДокумент12 страницCódigos InactivosJaime Añazco ManchegoОценок пока нет
- Rel Med de Ref 15-08-2013Документ184 страницыRel Med de Ref 15-08-2013Armando Cortes MendozaОценок пока нет
- Formas Farmacéuticas Sólidas de Administración de Fármacos 5Документ2 страницыFormas Farmacéuticas Sólidas de Administración de Fármacos 5edwinОценок пока нет
- TP-5 Fcdinamia II 2018Документ8 страницTP-5 Fcdinamia II 2018Federico AlessiОценок пока нет
- VII BIOFARMACIA - Plan 2014 AD APROBADOДокумент7 страницVII BIOFARMACIA - Plan 2014 AD APROBADORhenso Victor Albites CondoriОценок пока нет