Вам также может понравиться
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (120)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- Psychedelic Substances: The Little Book ofДокумент46 страницPsychedelic Substances: The Little Book ofpepe100% (2)
- Community Bank Priorities ReportДокумент34 страницыCommunity Bank Priorities ReportScottОценок пока нет
- Austen BioInnovation Institute General PresentationДокумент12 страницAusten BioInnovation Institute General PresentationScottОценок пока нет
- Resilience - Working Together To Reduce Disaster RiskДокумент32 страницыResilience - Working Together To Reduce Disaster RiskScottОценок пока нет
- Regulatory Challenges During Medical Device DevelopmentДокумент18 страницRegulatory Challenges During Medical Device DevelopmentScottОценок пока нет
- City Communications Plan ExampleДокумент19 страницCity Communications Plan ExampleScottОценок пока нет
- Our House RulesДокумент1 страницаOur House RulesScottОценок пока нет
- BEd GuidelinesДокумент84 страницыBEd GuidelinesshindochaОценок пока нет
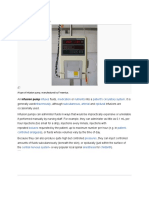
- Infusion Pump: Infuses Medication Nutrients Patient's Circulatory System Intravenously Subcutaneous Arterial EpiduralДокумент6 страницInfusion Pump: Infuses Medication Nutrients Patient's Circulatory System Intravenously Subcutaneous Arterial EpiduralpratonyОценок пока нет
- (New & Improved) TDEE Calculator (Insanely Accurate)Документ11 страниц(New & Improved) TDEE Calculator (Insanely Accurate)LaraОценок пока нет
- The Pathophysiologic Basis of Nuclear MedicineДокумент21 страницаThe Pathophysiologic Basis of Nuclear MedicineShazia FatimaОценок пока нет
- Epidemiology, Pathology, Clinical Features, and Diagnosis of Meningioma - UpToDateДокумент22 страницыEpidemiology, Pathology, Clinical Features, and Diagnosis of Meningioma - UpToDatewilson kores100% (1)
- Psychiatry EmergencyДокумент121 страницаPsychiatry EmergencyDr. Jayesh Patidar67% (6)
- Physical Layout PICUДокумент8 страницPhysical Layout PICUMeena Koushal0% (1)
- Vii. Nursing Care Plan: Assessment Nursing Diagnosis Inference Planning Intervention Rationale Evaluation SubjectiveДокумент2 страницыVii. Nursing Care Plan: Assessment Nursing Diagnosis Inference Planning Intervention Rationale Evaluation SubjectiveMezil NazarenoОценок пока нет
- Health Informatics in PakistanДокумент5 страницHealth Informatics in PakistanFatima Diane S. MondejarОценок пока нет
- Text For NoДокумент3 страницыText For NoMasita NazalinОценок пока нет
- Handwashinglessonsk 6thgradeДокумент25 страницHandwashinglessonsk 6thgradeWi MaryОценок пока нет
- Music and Health June 2020utasv PDFДокумент4 страницыMusic and Health June 2020utasv PDFPayneOgden54Оценок пока нет
- Drug Study Form 1 OBДокумент1 страницаDrug Study Form 1 OBJOSHUA NERZA LACANDOZEОценок пока нет
- MTP Act 1971Документ16 страницMTP Act 1971akash tiwariОценок пока нет
- Curriculum Vitae: Dyah Ika KrisnawatiДокумент5 страницCurriculum Vitae: Dyah Ika Krisnawatiwedus2Оценок пока нет
- Developed by The Teaching History of Pharmacy Committee of The History of Pharmacy SIG, 2017-18Документ75 страницDeveloped by The Teaching History of Pharmacy Committee of The History of Pharmacy SIG, 2017-18Durga MadhuriОценок пока нет
- Lack of Sleep Research PaperДокумент8 страницLack of Sleep Research Papergw1sj1yb100% (1)
- Ballad Scarce Resource Allocation Patient Notification LetterДокумент2 страницыBallad Scarce Resource Allocation Patient Notification LetterJosh Smith100% (1)
- Kidney PainДокумент2 страницыKidney PainAulia Abdillah RamadhanОценок пока нет
- NCM112 LP1 Rosales Reflective Writing 2Документ4 страницыNCM112 LP1 Rosales Reflective Writing 2Christine CalleyОценок пока нет
- Fourth Grading NotesДокумент70 страницFourth Grading NotesMini RinnОценок пока нет
- Disaster Management at Site and at HospitalДокумент21 страницаDisaster Management at Site and at HospitalAlmasОценок пока нет
- Cumbria and Lancs KCN Algorithm PDFДокумент18 страницCumbria and Lancs KCN Algorithm PDFratih83Оценок пока нет
- Review Combat Against COVID-19 Complications - Traditional Plant Lantana CamaraДокумент5 страницReview Combat Against COVID-19 Complications - Traditional Plant Lantana CamaraInternational Journal of Innovative Science and Research TechnologyОценок пока нет
- Level of Awareness About Sex Education Among First Year and Second Year Medical Technology StudentsДокумент57 страницLevel of Awareness About Sex Education Among First Year and Second Year Medical Technology Studentsbibibebebaby leeleeОценок пока нет
- CMS MLN Cognitive Assessment and Care Plan Services CPT Code 99483Документ4 страницыCMS MLN Cognitive Assessment and Care Plan Services CPT Code 99483Salomon GreenОценок пока нет
- Definition of GriefДокумент16 страницDefinition of GriefNaveen Eldose100% (1)
- Ro10 Bukidnon Provincial Medical Center Malaybalay CityДокумент37 страницRo10 Bukidnon Provincial Medical Center Malaybalay Citycaleb castilloОценок пока нет
- Blinded Study Synopsis FormatДокумент9 страницBlinded Study Synopsis FormatSok YeeОценок пока нет