Вам также может понравиться
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- Oral Desensitization To Penicillin For The Treatment of Pregnant Women With Syphilis - A Successful ProgramДокумент1 страницаOral Desensitization To Penicillin For The Treatment of Pregnant Women With Syphilis - A Successful ProgramDaniel Carvalho de AraújoОценок пока нет
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Changes in The Extracellular Matrix Due To Diabetes and Their Impact On Urinary ContinenceДокумент8 страницChanges in The Extracellular Matrix Due To Diabetes and Their Impact On Urinary ContinenceDaniel Carvalho de AraújoОценок пока нет
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Revista Brasileira de Ginecologia e Obstetrícia: ObjectiveДокумент2 страницыRevista Brasileira de Ginecologia e Obstetrícia: ObjectiveDaniel Carvalho de AraújoОценок пока нет
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- Ocular Changes During PregnancyДокумент1 страницаOcular Changes During PregnancyDaniel Carvalho de AraújoОценок пока нет
- Therapeutic Assessment of Vulvar Squamous Intraepithelial Lesions With CO2 Laser Vaporization in Immunosuppressed PatientsДокумент2 страницыTherapeutic Assessment of Vulvar Squamous Intraepithelial Lesions With CO2 Laser Vaporization in Immunosuppressed PatientsDaniel Carvalho de AraújoОценок пока нет
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- Body Mass Index Changes During Pregnancy and Perinatal Outcomes - A Cross-Sectional StudyДокумент1 страницаBody Mass Index Changes During Pregnancy and Perinatal Outcomes - A Cross-Sectional StudyDaniel Carvalho de AraújoОценок пока нет
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- Gestantes Brasileiras Precisam de Suplementação de IodoДокумент3 страницыGestantes Brasileiras Precisam de Suplementação de IodoDaniel Carvalho de AraújoОценок пока нет
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- Association Between Breast Arterial Calcifications and Cardiovascular Risk Factors in Menopausal WomenДокумент8 страницAssociation Between Breast Arterial Calcifications and Cardiovascular Risk Factors in Menopausal WomenDaniel Carvalho de AraújoОценок пока нет
- Revista Brasileira de Ginecologia e Obstetrícia: Tumor Borderline Do Ovário Localizado No Canal Inguinal: Relato de CasoДокумент7 страницRevista Brasileira de Ginecologia e Obstetrícia: Tumor Borderline Do Ovário Localizado No Canal Inguinal: Relato de CasoDaniel Carvalho de AraújoОценок пока нет
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Changes in The Extracellular Matrix Due To Diabetes and Their Impact On Urinary ContinenceДокумент8 страницChanges in The Extracellular Matrix Due To Diabetes and Their Impact On Urinary ContinenceDaniel Carvalho de AraújoОценок пока нет
- ValidationДокумент8 страницValidationDaniel Carvalho de AraújoОценок пока нет
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- Correction To In-Transit Development of Color Abnormalities in Turkey Breast Meat During Winter SeasonДокумент2 страницыCorrection To In-Transit Development of Color Abnormalities in Turkey Breast Meat During Winter SeasonDaniel Carvalho de AraújoОценок пока нет
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- MaternalДокумент8 страницMaternalDaniel Carvalho de AraújoОценок пока нет
- LevelsДокумент7 страницLevelsDaniel Carvalho de AraújoОценок пока нет
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- Contribution of SLC26A4 To The Molecular DiagnosisДокумент8 страницContribution of SLC26A4 To The Molecular DiagnosisDaniel Carvalho de AraújoОценок пока нет
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- Revista Brasileira de Ginecologia e ObstetríciaДокумент7 страницRevista Brasileira de Ginecologia e ObstetríciaDaniel Carvalho de AraújoОценок пока нет
- SerumДокумент8 страницSerumDaniel Carvalho de AraújoОценок пока нет
- A Synthetic Medium To Simulate Sugarcane MolassesДокумент13 страницA Synthetic Medium To Simulate Sugarcane MolassesDaniel Carvalho de AraújoОценок пока нет
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- Relevance of Serum Angiogenic Cytokines in Adult Patients With DermatomyositisДокумент8 страницRelevance of Serum Angiogenic Cytokines in Adult Patients With DermatomyositisDaniel Carvalho de AraújoОценок пока нет
- Airway and Parenchyma Immune Cells in Influenza A (H1N1) pdm09 Viral and Non-Viral Diffuse Alveolar DamageДокумент11 страницAirway and Parenchyma Immune Cells in Influenza A (H1N1) pdm09 Viral and Non-Viral Diffuse Alveolar DamageDaniel Carvalho de AraújoОценок пока нет
- Wien Effect of CD-ZN On Soil Clay Fraction and Their InteractionДокумент12 страницWien Effect of CD-ZN On Soil Clay Fraction and Their InteractionDaniel Carvalho de AraújoОценок пока нет
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- Peptidomic Investigation of Neoponera Villosa Venom by High-Resolution Mass Spectrometry - Seasonal and Nesting Habitat VariationsДокумент12 страницPeptidomic Investigation of Neoponera Villosa Venom by High-Resolution Mass Spectrometry - Seasonal and Nesting Habitat VariationsDaniel Carvalho de AraújoОценок пока нет
- Awareness and Current Knowledge of Breast CancerДокумент24 страницыAwareness and Current Knowledge of Breast CancerDaniel Carvalho de AraújoОценок пока нет
- Force Level of Small Diameter Nickel-Titanium Orthodontic Wires Ligated With Different MethodsДокумент8 страницForce Level of Small Diameter Nickel-Titanium Orthodontic Wires Ligated With Different MethodsDaniel Carvalho de AraújoОценок пока нет
- Building A Resistance To Ignition Testing Device For Sunglasses and Analysing Data - A Continuing Study For Sunglasses StandardsДокумент11 страницBuilding A Resistance To Ignition Testing Device For Sunglasses and Analysing Data - A Continuing Study For Sunglasses StandardsDaniel Carvalho de AraújoОценок пока нет
- The Specific and Combined Role of Domestic Violence and Mental Health Disorders During Pregnancy On New-Born HealthДокумент11 страницThe Specific and Combined Role of Domestic Violence and Mental Health Disorders During Pregnancy On New-Born HealthDaniel Carvalho de AraújoОценок пока нет
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- Building A Resistance To Ignition Testing DeviceДокумент11 страницBuilding A Resistance To Ignition Testing DeviceDaniel Carvalho de AraújoОценок пока нет
- Responsiveness of The Early Childhood Oral Health Impact Scale (ECOHIS) Is Related To Dental Treatment ComplexityДокумент10 страницResponsiveness of The Early Childhood Oral Health Impact Scale (ECOHIS) Is Related To Dental Treatment ComplexityDaniel Carvalho de AraújoОценок пока нет
- Responsiveness of The Early Childhood OralДокумент10 страницResponsiveness of The Early Childhood OralDaniel Carvalho de AraújoОценок пока нет
- Wing Variation in Culex Nigripalpus (Diptera - Culicidae) in Urban ParksДокумент9 страницWing Variation in Culex Nigripalpus (Diptera - Culicidae) in Urban ParksDaniel Carvalho de AraújoОценок пока нет
- MEBERC PhyChemДокумент122 страницыMEBERC PhyChemJames Ivan Palanas RotorОценок пока нет
- A1hex Lu6v8Документ24 страницыA1hex Lu6v8andremaxwelОценок пока нет
- Valves BasicsДокумент94 страницыValves BasicsSamuel Onyewuenyi100% (1)
- MCLДокумент4 страницыMCLDan CasuraoОценок пока нет
- Fluid Flow Postlab ReportДокумент14 страницFluid Flow Postlab Reportgracebrewster123Оценок пока нет
- Is 8147Документ170 страницIs 8147Asha JatalОценок пока нет
- l1 IntroductionДокумент20 страницl1 IntroductionCindy BergamoteОценок пока нет
- Final Quiz - Asme Pcc-2Документ15 страницFinal Quiz - Asme Pcc-2ajaysharma_100975% (4)
- RS6100004Документ52 страницыRS6100004e_readingОценок пока нет
- Torque and Rotation Quiz ReviewДокумент4 страницыTorque and Rotation Quiz ReviewMichelle ChenОценок пока нет
- Physical Chemistry IIДокумент70 страницPhysical Chemistry IIAyobami Akindele50% (2)
- SpaceCAD Model Rocket SoftwareДокумент7 страницSpaceCAD Model Rocket Softwareheric19886445Оценок пока нет
- University of Technology, JamaicaДокумент7 страницUniversity of Technology, JamaicaAthinaОценок пока нет
- MP3000 Controller Alarm and Warnings OverviewДокумент4 страницыMP3000 Controller Alarm and Warnings OverviewGomesОценок пока нет
- Design of Gentry GirderДокумент21 страницаDesign of Gentry GirderDebarshi SahooОценок пока нет
- Chemistry 1st Year Set-1 (English Medium) 2021 Guess PaperДокумент2 страницыChemistry 1st Year Set-1 (English Medium) 2021 Guess PaperPawan Kalyan JpkОценок пока нет
- Reductor SHB50-FДокумент28 страницReductor SHB50-FJaime Casas-corderoОценок пока нет
- Tech Guide D3SK 36-300 - 0503Документ20 страницTech Guide D3SK 36-300 - 0503saleh mohamedОценок пока нет
- A Parametric Study On Shoring Structures With Multi-Row Anchors in Layered SoilДокумент9 страницA Parametric Study On Shoring Structures With Multi-Row Anchors in Layered SoilJacky LeongОценок пока нет
- UntitledДокумент2 страницыUntitledJohnmarc Prince MontanoОценок пока нет
- Hybridization: DR Karimah Kassim CHM 475Документ34 страницыHybridization: DR Karimah Kassim CHM 475api-386303659Оценок пока нет
- Assignment - IДокумент4 страницыAssignment - IchritОценок пока нет
- 300 C SailДокумент2 страницы300 C Sailchandrabhushan kushwahaОценок пока нет
- Visual Examination of Welds - Welds 3-14Документ64 страницыVisual Examination of Welds - Welds 3-14carlos100% (1)
- Science 7 Lesson Plan Saturated - Unsaturated SolutionДокумент9 страницScience 7 Lesson Plan Saturated - Unsaturated SolutionKresha Lluisma100% (1)
- Inorganic Cha 4Документ21 страницаInorganic Cha 4Adugnaw BiksОценок пока нет
- Heat Exchanger For StudentsДокумент71 страницаHeat Exchanger For StudentsYasaswi NathОценок пока нет
- Construction and Working of A Babinet's CompensatorДокумент3 страницыConstruction and Working of A Babinet's CompensatorKhushal shendeОценок пока нет
- Surfactants For Use As CodispersantsДокумент10 страницSurfactants For Use As Codispersantsramitkatyal21881Оценок пока нет
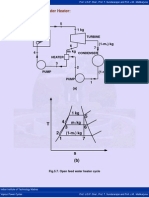
- OFWHДокумент2 страницыOFWHapi-3830954Оценок пока нет
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideОт EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuideОценок пока нет
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolОт EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolОценок пока нет
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactОт EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactРейтинг: 5 из 5 звезд5/5 (5)
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincОт EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincРейтинг: 3.5 из 5 звезд3.5/5 (137)
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeОт EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeРейтинг: 5 из 5 звезд5/5 (4)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeОт EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeРейтинг: 4 из 5 звезд4/5 (1)