Вам также может понравиться
- +50 Business Model Examples PDFДокумент119 страниц+50 Business Model Examples PDFKanjakha Pal50% (2)
- Success Story Uni HD - BASFДокумент1 страницаSuccess Story Uni HD - BASFKanjakha PalОценок пока нет
- Table 1 - List of EmployersДокумент4 страницыTable 1 - List of EmployersKanjakha PalОценок пока нет
- Guideline: Computational Interdisciplinary Graduate ProgramsДокумент16 страницGuideline: Computational Interdisciplinary Graduate ProgramsKanjakha PalОценок пока нет
- Soy Products GДокумент48 страницSoy Products GKanjakha PalОценок пока нет
- Global Top 50Документ4 страницыGlobal Top 50Kanjakha Pal100% (1)
- Icb 15 Sep 18Документ5 страницIcb 15 Sep 18123456789pppppОценок пока нет
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
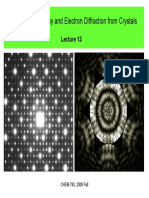
- Lecture 13 1-CHEM793 PDFДокумент30 страницLecture 13 1-CHEM793 PDFsanta ramirez lopezОценок пока нет
- Laporan Soil Investigation Pertamina PanjangДокумент60 страницLaporan Soil Investigation Pertamina PanjangAlex SudrajatОценок пока нет
- Experiment No. 3: University of The East - Caloocan College of Engineering Civil Engineering DepartmentДокумент6 страницExperiment No. 3: University of The East - Caloocan College of Engineering Civil Engineering DepartmentJHONRUSSEL ANTONIOОценок пока нет
- Sri Chaitanya IIT Academy., India.: Key Sheet PhysicsДокумент10 страницSri Chaitanya IIT Academy., India.: Key Sheet PhysicsCE-026 BharadwajaОценок пока нет
- Experiment 3: Determination of Lead in Anchovies by Cold Vapour Generation Atomic Absorption SpectrometryДокумент31 страницаExperiment 3: Determination of Lead in Anchovies by Cold Vapour Generation Atomic Absorption SpectrometrymanurihimalshaОценок пока нет
- C-SCP360H38B R410aДокумент8 страницC-SCP360H38B R410aRaúl RodríguezОценок пока нет
- Heat Exchanger Question - ANSДокумент7 страницHeat Exchanger Question - ANSBikas SahaОценок пока нет
- Vibration Analysis and Diagnostic GuideДокумент66 страницVibration Analysis and Diagnostic GuideAgung Budi Tri PrasetyoОценок пока нет
- SBT 200 Plant Ecologykenyatta UniversityДокумент85 страницSBT 200 Plant Ecologykenyatta Universityvincent onsaseОценок пока нет
- Uniheat - Transfer Thermal Unitst II Convective Heat Transfer Thermal UnitsДокумент33 страницыUniheat - Transfer Thermal Unitst II Convective Heat Transfer Thermal Unitsaerosanth100% (1)
- CH 7.4 - PPT - To Students - 25-3-2023Документ13 страницCH 7.4 - PPT - To Students - 25-3-2023jujuОценок пока нет
- SS Connection For "W" Shapes A572 Grade 50 Steel Is Used Whoes Properties Are As Below: Fy 345 Mpa, Fu 450 MpaДокумент31 страницаSS Connection For "W" Shapes A572 Grade 50 Steel Is Used Whoes Properties Are As Below: Fy 345 Mpa, Fu 450 MpaNats SantosОценок пока нет
- Properties and Characterization of Al-Al2O3 Composites PDFДокумент11 страницProperties and Characterization of Al-Al2O3 Composites PDFudaypattelaОценок пока нет
- Line Data: Piping Isometric DrawingsДокумент1 страницаLine Data: Piping Isometric Drawingsksat85Оценок пока нет
- Ascentis Express Hilic Guide Omi t412061Документ12 страницAscentis Express Hilic Guide Omi t412061Jaycer AsbyssОценок пока нет
- Safety ValvesДокумент8 страницSafety ValvesvenkeekuОценок пока нет
- Handbook of Pig MedicineДокумент14 страницHandbook of Pig MedicineSophorTra SopheakОценок пока нет
- Earth Science Module 5 AnswersheetДокумент2 страницыEarth Science Module 5 AnswersheetCorazon MARCIANOОценок пока нет
- CH - 12 ElectricityДокумент24 страницыCH - 12 ElectricityJency JohnsonОценок пока нет
- Calculations and Verifications of Shredding Chamber of Two-Shaft Shredder For Crushing of Concrete, Rubber, Plastic and WoodДокумент2 страницыCalculations and Verifications of Shredding Chamber of Two-Shaft Shredder For Crushing of Concrete, Rubber, Plastic and WoodEdosael KefyalewОценок пока нет
- Pinnacle Alloys E9018-M Code and Specification DataДокумент2 страницыPinnacle Alloys E9018-M Code and Specification DataPutra Panca WardhanaОценок пока нет
- Gate 2000 PDFДокумент14 страницGate 2000 PDFVammsy Manikanta SaiОценок пока нет
- NSS Phy Curriculum (Notes For Teachers)Документ30 страницNSS Phy Curriculum (Notes For Teachers)energy0124Оценок пока нет
- Maximizing Wet Scrubber PerformanceДокумент7 страницMaximizing Wet Scrubber PerformanceHESuarez100% (1)
- Newtons Laws and MotionДокумент11 страницNewtons Laws and Motionvictoria kairooОценок пока нет
- Plasma Orbital Expansion Electrons Water FrequencyДокумент140 страницPlasma Orbital Expansion Electrons Water FrequencyVincent J. Cataldi100% (1)
- Experiment 4: Work, Power and Energy Laboratory ReportДокумент4 страницыExperiment 4: Work, Power and Energy Laboratory ReportRamnuj Orecul SoralcОценок пока нет
- Accelerated Aging and Thermal-Mechanical Fatigue MДокумент27 страницAccelerated Aging and Thermal-Mechanical Fatigue MQUALITY LABORATORYОценок пока нет
- Aws D1.1welding Qualification.Документ10 страницAws D1.1welding Qualification.idealparrotОценок пока нет
- Header BowmanДокумент12 страницHeader BowmanMehrdad SakhaieОценок пока нет