Вам также может понравиться
- An Efficient, Optimized Synthesis of Fentanyl and Related AnalogsДокумент8 страницAn Efficient, Optimized Synthesis of Fentanyl and Related AnalogsJay MasonОценок пока нет
- A Synthesis of Tropinone PDFДокумент4 страницыA Synthesis of Tropinone PDFjustinldickeyОценок пока нет
- Determination of Synthesis Method of Ecstasy Based On The Basic ImpuritiesДокумент10 страницDetermination of Synthesis Method of Ecstasy Based On The Basic Impuritiesgeovani2100% (1)
- Codeine To MorphineДокумент2 страницыCodeine To MorphineBenjamin HartОценок пока нет
- Synthesis and Analgesic Properties of N-Substituted Trans-4a-Aryldecahydroisoquinolines - J. Med. Chem., 1988, 31 (3), PP 555-560Документ6 страницSynthesis and Analgesic Properties of N-Substituted Trans-4a-Aryldecahydroisoquinolines - J. Med. Chem., 1988, 31 (3), PP 555-560muopioidreceptor100% (1)
- A Rapid, High-Yield Conversion of Codeine To MorphineДокумент2 страницыA Rapid, High-Yield Conversion of Codeine To Morphinegeovani250% (2)
- ALKALOID With Some IsolationДокумент24 страницыALKALOID With Some Isolationapi-3742263100% (23)
- DPS 1-3-126 139 Potent FentДокумент14 страницDPS 1-3-126 139 Potent Fentlumik1234Оценок пока нет
- A Facile and Improved Synthesis of DesomorphineДокумент4 страницыA Facile and Improved Synthesis of DesomorphineAnonymous 4KaJRMОценок пока нет
- Andy Wong - Synthetic Opium: The Occurrence, Bioactivity, Biosynthesis and Synthesis of OxycodoneДокумент4 страницыAndy Wong - Synthetic Opium: The Occurrence, Bioactivity, Biosynthesis and Synthesis of OxycodonePoloGreen100% (1)
- Improved Procedure For The Preparation of 1 - (2-Phenethyl) - 4-PiperidoneДокумент5 страницImproved Procedure For The Preparation of 1 - (2-Phenethyl) - 4-PiperidonejesusОценок пока нет
- Morphine ChemistryДокумент19 страницMorphine ChemistryRam Babu TripathiОценок пока нет
- Synthesis of Piperidine - MGP Buffat - Tetrahedron, 2004, 60 (8), 1701-1729Документ29 страницSynthesis of Piperidine - MGP Buffat - Tetrahedron, 2004, 60 (8), 1701-1729muopioidreceptor100% (1)
- Submitted by Department of Chemistry, Imam Hossein University, Tehran, IRANДокумент4 страницыSubmitted by Department of Chemistry, Imam Hossein University, Tehran, IRANsinaОценок пока нет
- Effects of Bath Salts Drug MDPVДокумент21 страницаEffects of Bath Salts Drug MDPVOmar ZourobОценок пока нет
- Calcium Gluconate USP MonographДокумент4 страницыCalcium Gluconate USP Monographthe docsproviderОценок пока нет
- Opium 160604163652Документ14 страницOpium 160604163652roberto cabungcagОценок пока нет
- Synthesis ofДокумент2 страницыSynthesis ofHofman HofmannОценок пока нет
- Pseudoephedrine: 1. Synonyms CFR: Nist #Документ18 страницPseudoephedrine: 1. Synonyms CFR: Nist #Yuyun Saputri NingsihОценок пока нет
- The Chemistry of The Morphine Alkaloids Bentley (Oxford 1954) PDFДокумент452 страницыThe Chemistry of The Morphine Alkaloids Bentley (Oxford 1954) PDFJim Leonard100% (2)
- LSD Synthesis GuideДокумент12 страницLSD Synthesis GuideKyle Ryan100% (1)
- The Total Syntheses of Ring-A Substituted ErgolinesДокумент139 страницThe Total Syntheses of Ring-A Substituted ErgolinespmaldonatoОценок пока нет
- The Morphine AlkaloidsДокумент159 страницThe Morphine Alkaloidsboogiedee1100% (1)
- Omcl0204 - 0181 - Synthesis of MescalineДокумент10 страницOmcl0204 - 0181 - Synthesis of MescalineOleksandr StorcheusОценок пока нет
- Opiate - Chemistry.and - Metabolism BedfordДокумент5 страницOpiate - Chemistry.and - Metabolism BedfordSam SlaterОценок пока нет
- Development of Potent μ and δ Opioid Agonists with High Lipophilicity - J. Med. Chem., 2011, 54 (1), pp 382-386Документ5 страницDevelopment of Potent μ and δ Opioid Agonists with High Lipophilicity - J. Med. Chem., 2011, 54 (1), pp 382-386muopioidreceptorОценок пока нет
- Improved Bromination of 2C HДокумент3 страницыImproved Bromination of 2C HhappylmОценок пока нет
- Morphine Synthesis and Biosynthesis-An UpdateДокумент21 страницаMorphine Synthesis and Biosynthesis-An UpdatejaОценок пока нет
- Two-Step Protic Solvent-Catalyzed Reaction of Phenylethylamine With Methyl Acrylate (Organic Preparations and Procedures International, 2005, 37, 6, 579-584 10.1080@00304940509354990)Документ7 страницTwo-Step Protic Solvent-Catalyzed Reaction of Phenylethylamine With Methyl Acrylate (Organic Preparations and Procedures International, 2005, 37, 6, 579-584 10.1080@00304940509354990)DmitryОценок пока нет
- The Total Synthesis of Lysergic AcidДокумент28 страницThe Total Synthesis of Lysergic AcidFabio CavalcanteОценок пока нет
- Alkaloids: Alkaloids Are A Group of Naturally Occurring Chemical Compounds That Contain Mostly BasicДокумент13 страницAlkaloids: Alkaloids Are A Group of Naturally Occurring Chemical Compounds That Contain Mostly Basicgulshan araОценок пока нет
- Organic Chemistry Lab Manual SolubilityДокумент108 страницOrganic Chemistry Lab Manual SolubilityTri shiaОценок пока нет
- Sharma2018 Article The Search For The Next Euphoric NonДокумент17 страницSharma2018 Article The Search For The Next Euphoric NonDr. Ghulam FareedОценок пока нет
- Permissible Limit of Drinking WaterДокумент6 страницPermissible Limit of Drinking WaterronitОценок пока нет
- A Convenient Way To Synthesis of Analgesic TramadolДокумент1 страницаA Convenient Way To Synthesis of Analgesic TramadolFacundo BaróОценок пока нет
- Legal Chemistry: A Guide to the Detection of Poisons, Examination of Tea, Stains, Etc., as Applied to Chemical JurisprudenceОт EverandLegal Chemistry: A Guide to the Detection of Poisons, Examination of Tea, Stains, Etc., as Applied to Chemical JurisprudenceОценок пока нет
- Sintese Do 2c-bДокумент13 страницSintese Do 2c-bRF BragaОценок пока нет
- Cocaine Synthesis Shing Org Lett 2011Документ4 страницыCocaine Synthesis Shing Org Lett 2011Cody DunnОценок пока нет
- Alkaloidal AminesДокумент31 страницаAlkaloidal Aminesharishkumar kakrani100% (3)
- Recent Advances in The Chemistry of Oripavine & DerivativesABB - 2014072111275178Документ15 страницRecent Advances in The Chemistry of Oripavine & DerivativesABB - 2014072111275178Ji ChemОценок пока нет
- Preparation of Acetic Ester Morphine AlkaloidДокумент6 страницPreparation of Acetic Ester Morphine AlkaloidJaroslawkaczynskiОценок пока нет
- Short Communication - A Novel Synthesis of 3 4-Methylenedioxyphenyl-2-Propanone MDP2P From HelionalДокумент3 страницыShort Communication - A Novel Synthesis of 3 4-Methylenedioxyphenyl-2-Propanone MDP2P From HelionalMikel L.Оценок пока нет
- Enhanced Codeine and Morphine Production in SuspendedДокумент5 страницEnhanced Codeine and Morphine Production in SuspendedPádraig Ó ĊonġaileОценок пока нет
- A John Blacker - Mike T Williams - Pharmaceutical Process Development - Current Chemical and Engineering Challenges-Royal Society of Chemistry (2011)Документ375 страницA John Blacker - Mike T Williams - Pharmaceutical Process Development - Current Chemical and Engineering Challenges-Royal Society of Chemistry (2011)Nguyen Dang Binh ThanhОценок пока нет
- Advances in N- and O-Demethylation MethodsДокумент26 страницAdvances in N- and O-Demethylation MethodsAnonymous 4KaJRMОценок пока нет
- Guideline For The Assessment of Mosh Moah PDFДокумент52 страницыGuideline For The Assessment of Mosh Moah PDFSaid Toro Uribe100% (1)
- Morfin 2Документ6 страницMorfin 2Nova DhilaОценок пока нет
- Monographs On The Chemistry of Natural Products: Sir Bobbbt BobinsonДокумент452 страницыMonographs On The Chemistry of Natural Products: Sir Bobbbt BobinsonLasitarda BluesОценок пока нет
- Chemical Profiling of Heroin RecoveredДокумент6 страницChemical Profiling of Heroin RecoveredColo Volta100% (1)
- Trevor Sherwood - Morphine: Molecule in ReviewДокумент15 страницTrevor Sherwood - Morphine: Molecule in ReviewPoloGreenОценок пока нет
- Opioid Conversion ChartДокумент4 страницыOpioid Conversion ChartVanessa NicoleОценок пока нет
- Psyc1022 Topic 1Документ8 страницPsyc1022 Topic 1PatriciaОценок пока нет
- Marjolein Gabriëlle Klous - Pharmaceutical Development of Diacetylmorphine Preparations For Prescription To Opioid Dependent PatientsДокумент205 страницMarjolein Gabriëlle Klous - Pharmaceutical Development of Diacetylmorphine Preparations For Prescription To Opioid Dependent PatientsPoloGreenОценок пока нет
- The Preparation and Properties of CodeinoneДокумент3 страницыThe Preparation and Properties of Codeinonegeovani2100% (1)
- HL Paper3Документ18 страницHL Paper3charlesma123Оценок пока нет
- Piperidone AnalogsДокумент10 страницPiperidone AnalogssinaОценок пока нет
- Chemistry of Opium CompleteДокумент9 страницChemistry of Opium Completecarlitog782Оценок пока нет
- Levo SynДокумент7 страницLevo Synziggy862003100% (1)
- Mu Opioids and Their Receptors Evolution of A Concept - GW Pasternak - Pharmacol Rev (2013) - REVIEWДокумент61 страницаMu Opioids and Their Receptors Evolution of A Concept - GW Pasternak - Pharmacol Rev (2013) - REVIEWJonathan Berry100% (2)
- Opioids MorphineДокумент9 страницOpioids MorphineTueОценок пока нет
- Morphine Total SynthesisДокумент3 страницыMorphine Total SynthesisLeidy Tatiana Lievano RubioОценок пока нет
- Substitution Reactions of 2-Phenylsulphonyl Piperidines & 2-Phenylsulphonyl Pyrrolidines With Carbon Nucleophiles - Tetrahedron, 1991, 47 (7), 1311Документ18 страницSubstitution Reactions of 2-Phenylsulphonyl Piperidines & 2-Phenylsulphonyl Pyrrolidines With Carbon Nucleophiles - Tetrahedron, 1991, 47 (7), 1311muopioidreceptorОценок пока нет
- Newer Methods of Preparative Organic Chemistry V2От EverandNewer Methods of Preparative Organic Chemistry V2Wilhelm FoerstОценок пока нет
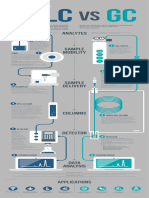
- HPLC Vs GCДокумент1 страницаHPLC Vs GCVinay PatelОценок пока нет
- GcmsДокумент3 страницыGcmsRista Susanti100% (1)
- Validated Spectroscopic Method For Estimation of Sumatriptan Succinate in Pure and From Tablet FormulationДокумент3 страницыValidated Spectroscopic Method For Estimation of Sumatriptan Succinate in Pure and From Tablet FormulationPrabhuОценок пока нет
- Class 11 - Chemistry - Organic Chemistry Some Basic PrinciplesДокумент30 страницClass 11 - Chemistry - Organic Chemistry Some Basic PrinciplesSachit GuptaОценок пока нет
- Journal of Pharmaceutical and Scientific Innovation: Lawsonia Inermis Linn. and Its Quantitative Analysis by HPTLCДокумент4 страницыJournal of Pharmaceutical and Scientific Innovation: Lawsonia Inermis Linn. and Its Quantitative Analysis by HPTLCnanoОценок пока нет
- Certificate of Analysis for Vitamin A Palmitate CRMДокумент6 страницCertificate of Analysis for Vitamin A Palmitate CRMAnonymous pCIauPOGОценок пока нет
- Isolation of A New Phlorotannin Fucodiphlorethol G From A Brown Alga ExtractДокумент3 страницыIsolation of A New Phlorotannin Fucodiphlorethol G From A Brown Alga ExtractlongchinОценок пока нет
- HPLC Guide: Principles, Applications and DetectorsДокумент30 страницHPLC Guide: Principles, Applications and DetectorsPatchiko HealmeОценок пока нет
- Agilent 1100MSD GettingStartedДокумент32 страницыAgilent 1100MSD GettingStartedMarine JolieОценок пока нет
- CHM510 Exp 5Документ12 страницCHM510 Exp 5NURANISAH NADIAH MOHD NIZAMОценок пока нет
- Additive 510 m14Документ5 страницAdditive 510 m14MohsenОценок пока нет
- Degradation-Emissions of Solvent (Buvik 2021) )Документ19 страницDegradation-Emissions of Solvent (Buvik 2021) )Wayne MonneryОценок пока нет
- 3-Phase Sparator Sizing (Vertical and Horizontal)Документ30 страниц3-Phase Sparator Sizing (Vertical and Horizontal)WickyОценок пока нет
- Food Analysis: Precision: A Measure of The Ability To Reproduce An Answer Between Determinations Performed by The SameДокумент11 страницFood Analysis: Precision: A Measure of The Ability To Reproduce An Answer Between Determinations Performed by The SameOLUWASEGUN K AfolabiОценок пока нет
- HPLC Columns Guide Br7614en MKДокумент122 страницыHPLC Columns Guide Br7614en MKFatih RahmawatiОценок пока нет
- Paracetamol MPДокумент3 страницыParacetamol MPMédi MenakuntimaОценок пока нет
- Trajan SGE Instrument Quick Pick Guide19095Документ24 страницыTrajan SGE Instrument Quick Pick Guide19095FeredОценок пока нет
- FA-derivatyzacja AChromДокумент13 страницFA-derivatyzacja AChromReza AzghadiОценок пока нет
- Jenke 2006Документ18 страницJenke 2006Ravi KanthОценок пока нет
- HPLCДокумент5 страницHPLCZed ZiumОценок пока нет
- Mechanistic Investigations On Multiproduct B-HimacДокумент45 страницMechanistic Investigations On Multiproduct B-HimacMuhammad Fakhrizal FahmiОценок пока нет
- MPharm - Pharmaceutics SyllabusДокумент21 страницаMPharm - Pharmaceutics Syllabussreeramraghu0% (1)
- What Is Quality ControlДокумент13 страницWhat Is Quality Controlchirag sabhayaОценок пока нет
- 1 - Analysis of Saturated and Aromatic HydrocarbonsДокумент17 страниц1 - Analysis of Saturated and Aromatic HydrocarbonsFaisal SaifОценок пока нет
- NF Monographs Benzalkonium Chloride SolutionДокумент3 страницыNF Monographs Benzalkonium Chloride Solutionsergio910113Оценок пока нет