Вам также может понравиться
- MCQ ERIA For KolegiumДокумент7 страницMCQ ERIA For KolegiumSylphana Astharica LawalataОценок пока нет
- MCQ ERIA For KolegiumДокумент7 страницMCQ ERIA For KolegiumSylphana Astharica LawalataОценок пока нет
- Kurva SuhuДокумент4 страницыKurva SuhuRendy YogaОценок пока нет
- Patognomonik CroupДокумент6 страницPatognomonik CroupRendy YogaОценок пока нет
- Annual Report - 2008 PDFДокумент246 страницAnnual Report - 2008 PDFRendy YogaОценок пока нет
- Kandungan Susu Formula PediatrikДокумент12 страницKandungan Susu Formula PediatrikRendy YogaОценок пока нет
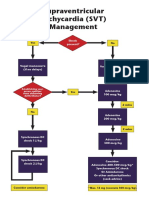
- Algorithms - SVT PDFДокумент1 страницаAlgorithms - SVT PDFjhontarroОценок пока нет
- Kandungan Susu Formula PediatrikДокумент12 страницKandungan Susu Formula PediatrikRendy YogaОценок пока нет
- TALKTOOLS - Feeding TherapyДокумент59 страницTALKTOOLS - Feeding TherapyRendy Yoga100% (1)
- Pubertas Prekok 2013Документ10 страницPubertas Prekok 2013Rendy YogaОценок пока нет
- Rendy Abstract NPM 1Документ1 страницаRendy Abstract NPM 1Rendy YogaОценок пока нет
- Abstrak Kardio RistaДокумент1 страницаAbstrak Kardio RistaRendy YogaОценок пока нет
- Case Report A 3 Week Old Baby With Disorder of Sex DevelopmentДокумент8 страницCase Report A 3 Week Old Baby With Disorder of Sex DevelopmentRendy YogaОценок пока нет
- Case Report A 3 Week Old Baby With Disorder of Sex DevelopmentДокумент11 страницCase Report A 3 Week Old Baby With Disorder of Sex DevelopmentRendy YogaОценок пока нет
- BGD Tutorial 2015 - Applied Embryology and TeratologyДокумент10 страницBGD Tutorial 2015 - Applied Embryology and TeratologyRendy YogaОценок пока нет
- Case Report A 3 Week Old Baby With Disorder of Sex DevelopmentДокумент8 страницCase Report A 3 Week Old Baby With Disorder of Sex DevelopmentRendy YogaОценок пока нет
- JI RendyДокумент6 страницJI RendyRendy YogaОценок пока нет
- JI RendyДокумент6 страницJI RendyRendy YogaОценок пока нет
- Ja2015 543928Документ6 страницJa2015 543928Rendy YogaОценок пока нет
- Technical Report PDFДокумент336 страницTechnical Report PDFKalina MassetОценок пока нет
- Fasilitas HotelДокумент16 страницFasilitas HotelRendy YogaОценок пока нет
- 1 5 Kamar Deluxe Executive Club Executive Suite 2 5 StandartДокумент3 страницы1 5 Kamar Deluxe Executive Club Executive Suite 2 5 StandartRendy YogaОценок пока нет
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- Developments in Prepress Technology (PDFDrive)Документ62 страницыDevelopments in Prepress Technology (PDFDrive)Sur VelanОценок пока нет
- Technical Manual: 110 125US 110M 135US 120 135UR 130 130LCNДокумент31 страницаTechnical Manual: 110 125US 110M 135US 120 135UR 130 130LCNKevin QuerubinОценок пока нет
- Sikkim Manipal MBA 1 SEM MB0038-Management Process and Organization Behavior-MQPДокумент15 страницSikkim Manipal MBA 1 SEM MB0038-Management Process and Organization Behavior-MQPHemant MeenaОценок пока нет
- SKF LGMT-2 Data SheetДокумент2 страницыSKF LGMT-2 Data SheetRahul SharmaОценок пока нет
- Carry Trade Calculator 1.54Документ3 страницыCarry Trade Calculator 1.54Gabriel RomanОценок пока нет
- Chapter 11 Walter Nicholson Microcenomic TheoryДокумент15 страницChapter 11 Walter Nicholson Microcenomic TheoryUmair QaziОценок пока нет
- Pie in The Sky 3Документ5 страницPie in The Sky 3arsi_yaarОценок пока нет
- On Applied EthicsДокумент34 страницыOn Applied Ethicsamanpatel78667% (3)
- 0901b8038042b661 PDFДокумент8 страниц0901b8038042b661 PDFWaqasAhmedОценок пока нет
- EMI-EMC - SHORT Q and AДокумент5 страницEMI-EMC - SHORT Q and AVENKAT PATILОценок пока нет
- Actus Reus and Mens Rea New MergedДокумент4 страницыActus Reus and Mens Rea New MergedHoorОценок пока нет
- CORDLESS PLUNGE SAW PTS 20-Li A1 PDFДокумент68 страницCORDLESS PLUNGE SAW PTS 20-Li A1 PDFΑλεξης ΝεοφυτουОценок пока нет
- RODECaster Pro II - DataSheet - V01 - 4Документ1 страницаRODECaster Pro II - DataSheet - V01 - 4lazlosОценок пока нет
- Two 2 Page Quality ManualДокумент2 страницыTwo 2 Page Quality Manualtony sОценок пока нет
- Strength and Microscale Properties of Bamboo FiberДокумент14 страницStrength and Microscale Properties of Bamboo FiberDm EerzaОценок пока нет
- Fin 3 - Exam1Документ12 страницFin 3 - Exam1DONNA MAE FUENTESОценок пока нет
- SEBI Circular Dated 22.08.2011 (Cirmirsd162011)Документ3 страницыSEBI Circular Dated 22.08.2011 (Cirmirsd162011)anantОценок пока нет
- Chapter 1.4Документ11 страницChapter 1.4Gie AndalОценок пока нет
- CW February 2013Документ60 страницCW February 2013Clint FosterОценок пока нет
- Tax Accounting Jones CH 4 HW SolutionsДокумент7 страницTax Accounting Jones CH 4 HW SolutionsLolaLaTraileraОценок пока нет
- IIBA Academic Membership Info-Sheet 2013Документ1 страницаIIBA Academic Membership Info-Sheet 2013civanusОценок пока нет
- Is 778 - Copper Alloy ValvesДокумент27 страницIs 778 - Copper Alloy ValvesMuthu KumaranОценок пока нет
- Harga H2H Pula-Paket Data - Saldo EWallet v31012022Документ10 страницHarga H2H Pula-Paket Data - Saldo EWallet v31012022lala cemiОценок пока нет
- S 101-01 - PDF - User Interface - Computer MonitorДокумент130 страницS 101-01 - PDF - User Interface - Computer Monitormborghesi1Оценок пока нет
- Ss1169 - Telecom Frameworx l1TMFДокумент65 страницSs1169 - Telecom Frameworx l1TMFPrince SinghОценок пока нет
- Dunham Bush Midwall Split R410a InverterДокумент2 страницыDunham Bush Midwall Split R410a InverterAgnaldo Caetano100% (1)
- Community-Based Monitoring System (CBMS) : An Overview: Celia M. ReyesДокумент28 страницCommunity-Based Monitoring System (CBMS) : An Overview: Celia M. ReyesDiane Rose LacenaОценок пока нет
- Pyro ShieldДокумент6 страницPyro Shieldmunim87Оценок пока нет
- Occupational Therapy in Mental HealthДокумент16 страницOccupational Therapy in Mental HealthjethasОценок пока нет
- Expected MCQs CompressedДокумент31 страницаExpected MCQs CompressedAdithya kesavОценок пока нет