Вам также может понравиться
- Capa - Boscon 2009Документ63 страницыCapa - Boscon 2009windli2014Оценок пока нет
- Assets TN101104SelectingParticleAbsorbtionLaserDiffractio 6 - tcm54 35808 PDFДокумент9 страницAssets TN101104SelectingParticleAbsorbtionLaserDiffractio 6 - tcm54 35808 PDFwindli2014Оценок пока нет
- Journal of Pharmaceutical and Biomedical AnalysisДокумент7 страницJournal of Pharmaceutical and Biomedical Analysiswindli2014Оценок пока нет
- Good Metrics Practice For Quality Management ReviewsДокумент2 страницыGood Metrics Practice For Quality Management Reviewswindli2014Оценок пока нет
- 无菌物品有效期影响因素的调查Документ4 страницы无菌物品有效期影响因素的调查windli2014Оценок пока нет
- Assets TN101104SelectingParticleAbsorbtionLaserDiffractio 6 - tcm54 35808 PDFДокумент9 страницAssets TN101104SelectingParticleAbsorbtionLaserDiffractio 6 - tcm54 35808 PDFwindli2014Оценок пока нет
- Assets AN101104RelatingPaintGlossToParticleSize 6 Tcm54 35468Документ8 страницAssets AN101104RelatingPaintGlossToParticleSize 6 Tcm54 35468windli2014Оценок пока нет
- 关于数据完整性和审计追踪审核的当前问题和解答Документ4 страницы关于数据完整性和审计追踪审核的当前问题和解答windli2014Оценок пока нет
- Multifactor Non-Linear Modeling For Accelerated Stability Analysis and PredictionДокумент9 страницMultifactor Non-Linear Modeling For Accelerated Stability Analysis and Predictionwindli2014Оценок пока нет
- Discussion of Process Enlargement Validation: Pharmaceutical & Engineering Design 2012 33Документ5 страницDiscussion of Process Enlargement Validation: Pharmaceutical & Engineering Design 2012 33windli2014Оценок пока нет
- FDA与ISPE关于质量量度的试点计划Документ14 страницFDA与ISPE关于质量量度的试点计划windli2014Оценок пока нет
- 拜耳 在线质量分析 PDFДокумент47 страниц拜耳 在线质量分析 PDFwindli2014Оценок пока нет
- How Accurate Are Your DilutionsДокумент5 страницHow Accurate Are Your Dilutionswindli2014Оценок пока нет
- Job Description: 附件 1, SOP-1-0044,版本 001 Attachment 1, SOP-1-0044,Ver 001Документ6 страницJob Description: 附件 1, SOP-1-0044,版本 001 Attachment 1, SOP-1-0044,Ver 001windli2014Оценок пока нет
- ECA Integrity LabData 2015Документ6 страницECA Integrity LabData 2015windli2014Оценок пока нет
- Efpia Comments Gdle Chemistry Active Substances Final-UpdatedДокумент34 страницыEfpia Comments Gdle Chemistry Active Substances Final-Updatedwindli2014Оценок пока нет
- 稳定性Документ31 страница稳定性windli2014Оценок пока нет
- Questions and Answers On The 'Guideline On The Limits of Genotoxic Impurities'Документ6 страницQuestions and Answers On The 'Guideline On The Limits of Genotoxic Impurities'windli2014Оценок пока нет
- 13 GLP Data Integrity DraftДокумент13 страниц13 GLP Data Integrity Draftwindli2014Оценок пока нет
- Trends in Enhancing API SolubilityДокумент5 страницTrends in Enhancing API Solubilitywindli2014Оценок пока нет
- Ombination Roducts: I CGMP RДокумент6 страницOmbination Roducts: I CGMP Rvijayns_250355172Оценок пока нет
- A Review of Statistical Outlier MethodsДокумент4 страницыA Review of Statistical Outlier Methodswindli2014Оценок пока нет
- 1-7 Specification WHOДокумент46 страниц1-7 Specification WHOelektron2010Оценок пока нет
- GMP Requirements For Certificates of AnalysisДокумент1 страницаGMP Requirements For Certificates of AnalysisMina Maher MikhailОценок пока нет
- Five Step Strategy PDFДокумент1 страницаFive Step Strategy PDFwindli2014Оценок пока нет
- Eca新闻:Eu委员会签发2份指南终版:辅料gmp和原料药gdpДокумент2 страницыEca新闻:Eu委员会签发2份指南终版:辅料gmp和原料药gdpwindli2014Оценок пока нет
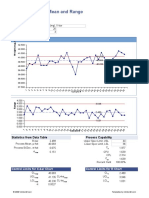
- Control Chart For Mean and Range: Quality CharacteristicДокумент3 страницыControl Chart For Mean and Range: Quality Characteristicwindli2014Оценок пока нет
- A Review On Quality by DesignДокумент8 страницA Review On Quality by Designwindli2014Оценок пока нет
- HPLC CalculatorДокумент16 страницHPLC Calculatorwindli2014Оценок пока нет
- 201308 Fda指南:Anda:原料药和制剂稳定性试验问答Документ32 страницы201308 Fda指南:Anda:原料药和制剂稳定性试验问答windli2014Оценок пока нет
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- Alinity Rubella IgGДокумент8 страницAlinity Rubella IgGLoloОценок пока нет
- Defic I Enc I A MultipleДокумент16 страницDefic I Enc I A MultipleEnrique GonzalvusОценок пока нет
- Monobind Reagent CatalogДокумент40 страницMonobind Reagent Catalogthuy dinhОценок пока нет
- Mabs Atezolimumab Flyer - KRIBIOLISA Atezolizumab / Anti-Atezolizumab ELISAДокумент2 страницыMabs Atezolimumab Flyer - KRIBIOLISA Atezolizumab / Anti-Atezolizumab ELISAKRISHGEN BIOSYSTEMSОценок пока нет
- Insert - Elecsys Free PSA.08828601500.V3.EnДокумент4 страницыInsert - Elecsys Free PSA.08828601500.V3.Entawfiq MohammadОценок пока нет
- BrochureДокумент2 страницыBrochurejoydeepsen06Оценок пока нет
- C-Peptide 03724875001 - enДокумент4 страницыC-Peptide 03724875001 - enModestusОценок пока нет
- Doping DetectionДокумент7 страницDoping DetectionPurdieparkerОценок пока нет
- Cobas E411 Sell SheetДокумент2 страницыCobas E411 Sell Sheetjalalnoureddeen100% (1)
- BT 505Документ4 страницыBT 505Hina MurtazaОценок пока нет
- Immunochemical Methods in The Clinical Laboratory: Roger L. Bertholf, PH.D., DABCC Mark A. Bowman, PH.D., MT (ASCP)Документ85 страницImmunochemical Methods in The Clinical Laboratory: Roger L. Bertholf, PH.D., DABCC Mark A. Bowman, PH.D., MT (ASCP)Milagros CamachoОценок пока нет
- Antigen and Antibody Reaction FinalДокумент77 страницAntigen and Antibody Reaction FinalSharma MukeshОценок пока нет
- Accre 8 BrochureДокумент2 страницыAccre 8 Brochuretingchen6688Оценок пока нет
- Prostate Specific Antigen (PSA) : Enzyme Immunoassay Test Kit Catalog Number: 10109Документ4 страницыProstate Specific Antigen (PSA) : Enzyme Immunoassay Test Kit Catalog Number: 10109yousrazeidan1979Оценок пока нет
- 8825-300 HCG-XR AccuBind ELISA Rev 3Документ2 страницы8825-300 HCG-XR AccuBind ELISA Rev 3petertrungОценок пока нет
- Fundamental Medical Science I Final Report (Proteomic & BSL)Документ12 страницFundamental Medical Science I Final Report (Proteomic & BSL)Vivian VallenciaОценок пока нет
- Hormones AnalysisДокумент41 страницаHormones AnalysisSULEIMAN OMAR100% (5)
- Architect Ci4100Документ7 страницArchitect Ci4100SantiagoAF50% (2)
- Cobas e 411 Leaflet enДокумент10 страницCobas e 411 Leaflet entomkuss1100% (2)
- 25-OHVD ManualДокумент2 страницы25-OHVD ManualOlga RodriguezОценок пока нет
- 099 Vitamin B12-IFU-V3.06-en-EUДокумент4 страницы099 Vitamin B12-IFU-V3.06-en-EUMohamed BoulkadidОценок пока нет
- Immunoassays: Fundamental Questions For An Analytical ChemistДокумент25 страницImmunoassays: Fundamental Questions For An Analytical ChemistDea Maylika SopiyantiОценок пока нет
- Navigating The Pandemic Response Life Cycle: Molecular Diagnostics and Immunoassays in The Context of COVID-19 ManagementДокумент19 страницNavigating The Pandemic Response Life Cycle: Molecular Diagnostics and Immunoassays in The Context of COVID-19 ManagementdebasreenitaОценок пока нет
- Immuno SeroДокумент80 страницImmuno SeroDocAxi Maximo Jr AxibalОценок пока нет
- Enzyme Linked Immunosorbent Assay For The Quantitative/qualitative Analysis of Plant Secondary MetabolitesДокумент11 страницEnzyme Linked Immunosorbent Assay For The Quantitative/qualitative Analysis of Plant Secondary MetabolitesIrfan MuhammadОценок пока нет
- Therapeutic Drug Monitoring: Saeed Alqahtani, Pharmd, PHDДокумент78 страницTherapeutic Drug Monitoring: Saeed Alqahtani, Pharmd, PHDAnonymous hF5zAdvwCC50% (2)
- Module 6 Post Lab DiscussionДокумент2 страницыModule 6 Post Lab DiscussionCiara PamonagОценок пока нет
- Bio-RadTechNote2861 Principles of Curve FittingДокумент4 страницыBio-RadTechNote2861 Principles of Curve Fittingyumyum9Оценок пока нет
- Drug Testing in Child WelfareДокумент49 страницDrug Testing in Child WelfareLOLA100% (1)
- ELISAДокумент3 страницыELISAAhmad ShahОценок пока нет