Вам также может понравиться
- Meier 2018 PostprintДокумент41 страницаMeier 2018 PostprintBehruz ArghavaniОценок пока нет
- 2D Multiphysics CFD Modeling of An ARP Carbothermic ReactorДокумент6 страниц2D Multiphysics CFD Modeling of An ARP Carbothermic ReactorDimitrios_Gerogiorgis4979Оценок пока нет
- Magnetocaloric Effect and Magnetic Cooling Near A Field-Induced Quantum-Critical PointДокумент21 страницаMagnetocaloric Effect and Magnetic Cooling Near A Field-Induced Quantum-Critical PointMirza MesanovicОценок пока нет
- An Enhanced Simulation Model For Building Envelopes With Phase Change MaterialsДокумент10 страницAn Enhanced Simulation Model For Building Envelopes With Phase Change MaterialsRam RamisettiОценок пока нет
- Identifying Heat and Mass Transfer Characteristics of Metal Hydride Reactor During Adsorption: Improved Formulation About Parameter AnalysisДокумент10 страницIdentifying Heat and Mass Transfer Characteristics of Metal Hydride Reactor During Adsorption: Improved Formulation About Parameter AnalysisjehadyamОценок пока нет
- 0035 001X RMF 67 01 114Документ9 страниц0035 001X RMF 67 01 114Adnane KinaniОценок пока нет
- tmpB8EA TMPДокумент8 страницtmpB8EA TMPFrontiersОценок пока нет
- Thermophysical Properties of Liquid Iron: Hlternatiomd Jourmd (" Thermophysics. Vol. 15. No. 6. 1994Документ9 страницThermophysical Properties of Liquid Iron: Hlternatiomd Jourmd (" Thermophysics. Vol. 15. No. 6. 1994Jose Velasquez TeranОценок пока нет
- A Mathematical Model of Gas Tungsten Arc Welding Considering The Cathode and The Free Surface of The Weld PooДокумент8 страницA Mathematical Model of Gas Tungsten Arc Welding Considering The Cathode and The Free Surface of The Weld Poohoangle1Оценок пока нет
- Paper Id 124Документ6 страницPaper Id 124kabeermuthuОценок пока нет
- Thermophysical Measurements On Liquid Iron and Nickel: High Temperatures-High Pressures January 1987Документ13 страницThermophysical Measurements On Liquid Iron and Nickel: High Temperatures-High Pressures January 1987Jose Velasquez TeranОценок пока нет
- Thermodynamic Properties of Minerals (Short Article) - A. Navrotsky (1995) WWДокумент11 страницThermodynamic Properties of Minerals (Short Article) - A. Navrotsky (1995) WWmonicaingenieraОценок пока нет
- TetratainiteДокумент11 страницTetratainitedpicc8Оценок пока нет
- Electrothermal Analysis OfElectric Resistance Spot Welding Processes by A 3-D Finite ElementДокумент6 страницElectrothermal Analysis OfElectric Resistance Spot Welding Processes by A 3-D Finite ElementBharadwajaChennupatiОценок пока нет
- Finite Element Analysis of Heat Flow inДокумент5 страницFinite Element Analysis of Heat Flow inWilly Villa SalsavilcaОценок пока нет
- Liu2004 PDFДокумент9 страницLiu2004 PDFLỢI NGUYỄN CÔNGОценок пока нет
- Integrated Multiphysics and Computational Fluid Dynamics Modeling of A Carbothermic Aluminium ReactorДокумент6 страницIntegrated Multiphysics and Computational Fluid Dynamics Modeling of A Carbothermic Aluminium ReactorOctaviano MichinelОценок пока нет
- Thermodynamics of Magnetic Refrigeration: Andrej Kitanovski, Peter W. EgolfДокумент19 страницThermodynamics of Magnetic Refrigeration: Andrej Kitanovski, Peter W. EgolfScott WaltonОценок пока нет
- The Thermodynamic Properties of Platinum: by J. W. ArblasterДокумент9 страницThe Thermodynamic Properties of Platinum: by J. W. ArblasterEmmanuel PlazaОценок пока нет
- Modeling of Radiation Intensity in An EAFДокумент6 страницModeling of Radiation Intensity in An EAFmehran1364Оценок пока нет
- Energies: Numerical Study On Thermal Hydraulic Performance of Supercritical LNG in Zigzag-Type Channel PchesДокумент19 страницEnergies: Numerical Study On Thermal Hydraulic Performance of Supercritical LNG in Zigzag-Type Channel PchesCrisMadiRodríguezBarríaОценок пока нет
- Energies: Numerical Study On Thermal Hydraulic Performance of Supercritical LNG in Zigzag-Type Channel PchesДокумент19 страницEnergies: Numerical Study On Thermal Hydraulic Performance of Supercritical LNG in Zigzag-Type Channel PchesSyed Anas SohailОценок пока нет
- arxScaleThermalConductivity BeygelzimerДокумент9 страницarxScaleThermalConductivity BeygelzimerЭммануил БикезинОценок пока нет
- Computational ThermodynamicsДокумент30 страницComputational ThermodynamicsRezzaPutraSamudraОценок пока нет
- Stationary and Transient Heat Conduction in A Non Homogeneous MaterialДокумент4 страницыStationary and Transient Heat Conduction in A Non Homogeneous MaterialVeronica ArevaloОценок пока нет
- Carpenter p2-22 98Документ21 страницаCarpenter p2-22 98mauricio rojas alvarezОценок пока нет
- Hydrodynamic Model For The Plasma-Gas Flow in A Cutting Torch NozzleДокумент7 страницHydrodynamic Model For The Plasma-Gas Flow in A Cutting Torch NozzleWilliam MasterОценок пока нет
- A Study On Numerical Analysis of The Resistance Spot Welding ProcessДокумент6 страницA Study On Numerical Analysis of The Resistance Spot Welding Processkiran_wakchaureОценок пока нет
- Thermophysical Properties of Containerless Liquid Iron Up To 2500 KДокумент10 страницThermophysical Properties of Containerless Liquid Iron Up To 2500 KJose Velasquez TeranОценок пока нет
- Research On Induction Heating - A ReviewДокумент4 страницыResearch On Induction Heating - A ReviewATSОценок пока нет
- A Novel Effective Medium Theory For Modelling The Thermal Conductivity of Porous MaterialsДокумент4 страницыA Novel Effective Medium Theory For Modelling The Thermal Conductivity of Porous Materialsmehmet8765Оценок пока нет
- Emanuela Giuffre and Franz Saija - Melting Line of Krypton in Extreme Thermodynamic RegimesДокумент10 страницEmanuela Giuffre and Franz Saija - Melting Line of Krypton in Extreme Thermodynamic RegimesDrebuioОценок пока нет
- Thermal Stability and Performance Data SmCo PPMДокумент8 страницThermal Stability and Performance Data SmCo PPMelectronenergyОценок пока нет
- Author's Accepted Manuscript: Journal of Physical and Chemistry of SolidsДокумент27 страницAuthor's Accepted Manuscript: Journal of Physical and Chemistry of SolidsJay Bee SharmaОценок пока нет
- Chapter - VДокумент24 страницыChapter - VKamel FedaouiОценок пока нет
- Experimental and Numerical Study On Temperature Control Performance of Phase Change Material Heat SinkДокумент33 страницыExperimental and Numerical Study On Temperature Control Performance of Phase Change Material Heat SinkManojkumar PОценок пока нет
- 5000 Grados PDFДокумент9 страниц5000 Grados PDFJose Velasquez TeranОценок пока нет
- Guevara & Irons (2011) Part IДокумент12 страницGuevara & Irons (2011) Part IaarondenboerОценок пока нет
- Rasouli Yazdi1998Документ11 страницRasouli Yazdi1998Antonio Alonso Diaz ArriagaОценок пока нет
- Large Magnetic Entropy Change Near Room Temperature in Antiperovskite Sncmn3Документ6 страницLarge Magnetic Entropy Change Near Room Temperature in Antiperovskite Sncmn3Evelyn TrianaОценок пока нет
- 5 1033 PDFДокумент12 страниц5 1033 PDFtahera aqeelОценок пока нет
- Gibbs Free Energy & Size Temperature Phase Diagram of HFNPS, Xiong Et Al., 2011Документ5 страницGibbs Free Energy & Size Temperature Phase Diagram of HFNPS, Xiong Et Al., 2011deryhermawanОценок пока нет
- Mathematical Modelling of Steel QuenchingДокумент6 страницMathematical Modelling of Steel Quenchingmanashree02Оценок пока нет
- Formal Kinetic Analysis of PVC Thermal Degradation: J. Blazevska-Gilev, D. SpaseskaДокумент4 страницыFormal Kinetic Analysis of PVC Thermal Degradation: J. Blazevska-Gilev, D. SpaseskaTrinh Quang ThanhОценок пока нет
- Review of The Magnetocaloric Effect in Manganite MaterialsДокумент16 страницReview of The Magnetocaloric Effect in Manganite MaterialsDhaval RanavasiyaОценок пока нет
- PH2610 2001aaaaaaaaaaaaaaДокумент6 страницPH2610 2001aaaaaaaaaaaaaabbteenagerОценок пока нет
- Groulx PaperДокумент7 страницGroulx PaperNook ThanatpornОценок пока нет
- Phase Diagrams: The Beginning of WisdomДокумент26 страницPhase Diagrams: The Beginning of WisdomOlga Sandoval RomeroОценок пока нет
- The Evaluation of Temperature Jump Distances and Thermal Accommodation CoefficientsДокумент10 страницThe Evaluation of Temperature Jump Distances and Thermal Accommodation CoefficientsMariah SmithОценок пока нет
- Heat Transfer Phenomena in A Solid Oxide Fuel Cell: An Analytical ApproachДокумент8 страницHeat Transfer Phenomena in A Solid Oxide Fuel Cell: An Analytical Approachpapillon tubaОценок пока нет
- MHD Simulation of Moving Arcs: 1-4244-0914-4/07/$25.00 ©2007 IEEE. 1013Документ5 страницMHD Simulation of Moving Arcs: 1-4244-0914-4/07/$25.00 ©2007 IEEE. 1013Felix GamarraОценок пока нет
- CALPHAD 36 (2012) 16-22: Critical Assessment: Martensite-Start Temperature for the γ → ε TransformationДокумент17 страницCALPHAD 36 (2012) 16-22: Critical Assessment: Martensite-Start Temperature for the γ → ε TransformationGanesh PMОценок пока нет
- Modeling and Experimental Verification of Martensitic Transformation Plastic Behavior in Carbon Steel For Quenching ProcessДокумент5 страницModeling and Experimental Verification of Martensitic Transformation Plastic Behavior in Carbon Steel For Quenching ProcessDanish KhanОценок пока нет
- 1 s2.0 S0925838813026832 MainДокумент11 страниц1 s2.0 S0925838813026832 Mainabdul basitОценок пока нет
- Steady State Temperature Distribution of Cast Resin Dry Type Transformer Based On New Thermal Model Using Finite Element MethodДокумент5 страницSteady State Temperature Distribution of Cast Resin Dry Type Transformer Based On New Thermal Model Using Finite Element MethodAnonymous sAmJfcVОценок пока нет
- Cryogenic Thermoelectric Cooler With A Passive BranchДокумент7 страницCryogenic Thermoelectric Cooler With A Passive BranchcharlesxdomОценок пока нет
- Paper Basico 1Документ12 страницPaper Basico 1Nacho Delgado FerreiroОценок пока нет
- Thermal Properties of Polymer/particle Composites at Low TemperaturesДокумент4 страницыThermal Properties of Polymer/particle Composites at Low TemperaturesSiva BhaskarОценок пока нет
- JunoCam Junos Outreach CameraДокумент32 страницыJunoCam Junos Outreach CameraJose Velasquez TeranОценок пока нет
- Thermophysical Measurements On Liquid Iron and Nickel: High Temperatures-High Pressures January 1987Документ13 страницThermophysical Measurements On Liquid Iron and Nickel: High Temperatures-High Pressures January 1987Jose Velasquez TeranОценок пока нет
- SodaPDF-converted - (English (Auto-Generated) ) The Attributes of God - Session 1 - Steve Lawson (DownSubДокумент27 страницSodaPDF-converted - (English (Auto-Generated) ) The Attributes of God - Session 1 - Steve Lawson (DownSubJose Velasquez TeranОценок пока нет
- An Introduction To Iron: ApplicationsДокумент2 страницыAn Introduction To Iron: ApplicationsJose Velasquez TeranОценок пока нет
- 5000 Grados PDFДокумент9 страниц5000 Grados PDFJose Velasquez TeranОценок пока нет
- Thermal Stresses in A Layered Plate: Created in COMSOL Multiphysics 5.4Документ12 страницThermal Stresses in A Layered Plate: Created in COMSOL Multiphysics 5.4Jose Velasquez TeranОценок пока нет
- Estimation of Effect of Process Parameters On Temperature, Thermal and Residual Stresses in Edmed Aisi D2 Steel ComponentsДокумент6 страницEstimation of Effect of Process Parameters On Temperature, Thermal and Residual Stresses in Edmed Aisi D2 Steel ComponentsJose Velasquez TeranОценок пока нет
- Christian Apologetics PHIL5301 New Orleans Baptist Theological Seminary January 7-11, 2019 Robert B. Stewart Office: Dodd 112, Extension #3245Документ6 страницChristian Apologetics PHIL5301 New Orleans Baptist Theological Seminary January 7-11, 2019 Robert B. Stewart Office: Dodd 112, Extension #3245Jose Velasquez TeranОценок пока нет
- BRACKДокумент1 страницаBRACKJose Velasquez TeranОценок пока нет
- UNIT 8-14 Review Audio PDFДокумент2 страницыUNIT 8-14 Review Audio PDFJose Velasquez TeranОценок пока нет
- Poster AERES MEB Muanalyses PDFДокумент1 страницаPoster AERES MEB Muanalyses PDFJose Velasquez TeranОценок пока нет
- Comparison of Dynamic Mechanical Properties Between Pure Iron (BCC) and Fe-30Mn-3Si-4Al TWIP Steel (FCC)Документ11 страницComparison of Dynamic Mechanical Properties Between Pure Iron (BCC) and Fe-30Mn-3Si-4Al TWIP Steel (FCC)Jose Velasquez TeranОценок пока нет
- Some The Temperature Range: Thermal ANDДокумент10 страницSome The Temperature Range: Thermal ANDJose Velasquez TeranОценок пока нет
- Thermophysical Properties of Liquid Iron: Hlternatiomd Jourmd (" Thermophysics. Vol. 15. No. 6. 1994Документ9 страницThermophysical Properties of Liquid Iron: Hlternatiomd Jourmd (" Thermophysics. Vol. 15. No. 6. 1994Jose Velasquez TeranОценок пока нет
- Dibitonto1989 PDFДокумент10 страницDibitonto1989 PDFJose Velasquez TeranОценок пока нет
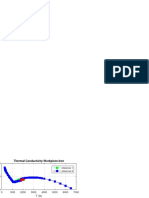
- Thermal Conductivity-Workpiece-Iron: Reference 1 Reference 2Документ1 страницаThermal Conductivity-Workpiece-Iron: Reference 1 Reference 2Jose Velasquez TeranОценок пока нет
- White 1994Документ11 страницWhite 1994Jose Velasquez TeranОценок пока нет
- Thermophysical Properties of Containerless Liquid Iron Up To 2500 KДокумент10 страницThermophysical Properties of Containerless Liquid Iron Up To 2500 KJose Velasquez TeranОценок пока нет
- Thermophysical Properties: Iron and SteelДокумент2 страницыThermophysical Properties: Iron and SteelJose Velasquez TeranОценок пока нет
- The Melodious Music Scale BookДокумент62 страницыThe Melodious Music Scale BookJose Velasquez Teran100% (3)
- KeywordsДокумент1 страницаKeywordsJose Velasquez TeranОценок пока нет
- Engineering Mechanics: (7th Edition)Документ3 страницыEngineering Mechanics: (7th Edition)Hassanein HeidarОценок пока нет
- Pairsof Angles LinesДокумент16 страницPairsof Angles LinesSero ArtsОценок пока нет
- Mathematics 3Документ142 страницыMathematics 3fafaОценок пока нет
- Taleb - Against VaRДокумент4 страницыTaleb - Against VaRShyamal VermaОценок пока нет
- Jackson 1.4 Homework Problem SolutionДокумент4 страницыJackson 1.4 Homework Problem SolutionJhonОценок пока нет
- Operational Objectives DefinitionДокумент1 страницаOperational Objectives DefinitionZven BlackОценок пока нет
- Chapter 1Документ24 страницыChapter 1Eunice SiervoОценок пока нет
- Section 1-1: Integration by Parts: SubstitutionsДокумент13 страницSection 1-1: Integration by Parts: SubstitutionsTony111Оценок пока нет
- Mathematical StudiesДокумент3 страницыMathematical StudiesEddy R. VélezОценок пока нет
- Bahan CR 2Документ19 страницBahan CR 2Exty RikaОценок пока нет
- All in One Prof EdДокумент65 страницAll in One Prof EdJhanina Apdua100% (1)
- Mellman - The Transverse Motion of Solids in Rotating Cylinders-Forms of Motion and Transitions BehaviorДокумент20 страницMellman - The Transverse Motion of Solids in Rotating Cylinders-Forms of Motion and Transitions BehaviorBrenno FerreiraОценок пока нет
- Brownian Motion: Langevin EquationДокумент14 страницBrownian Motion: Langevin Equationctasaltin100% (2)
- Leary 2001 Introduction To Behavioral Research Methods Capitol 9Документ25 страницLeary 2001 Introduction To Behavioral Research Methods Capitol 9Diana AndriescuОценок пока нет
- Precalculus ch6 ReviewДокумент2 страницыPrecalculus ch6 Reviewapi-213604106Оценок пока нет
- Exp 8Документ3 страницыExp 8Kurl Vincent GamboaОценок пока нет
- Lead4ward IQ ToolДокумент14 страницLead4ward IQ TooldowntownameerОценок пока нет
- The Role of Statistics in EngineeringДокумент42 страницыThe Role of Statistics in EngineeringMohammed Abushammala50% (2)
- Trigonometry Level 1 Test: Committed To Excellence in EducationДокумент2 страницыTrigonometry Level 1 Test: Committed To Excellence in EducationShreyas BansalОценок пока нет
- Post Laboratory Report - Response of First and Second Order SystemsДокумент13 страницPost Laboratory Report - Response of First and Second Order SystemsJulia GimenezОценок пока нет
- ADA104915 HEC BeardДокумент17 страницADA104915 HEC BeardVageesha Shantha Veerabhadra SwamyОценок пока нет
- Aqa MM1B QP Jun13 PDFДокумент24 страницыAqa MM1B QP Jun13 PDFastargroupОценок пока нет
- At114 Audit Sampling PDF FreeДокумент8 страницAt114 Audit Sampling PDF FreeKaila SalemОценок пока нет
- GDT Day1Документ119 страницGDT Day1Narendrareddy RamireddyОценок пока нет
- Analytic Statements Empirically Verifiable Statements: The Verification PrincipleДокумент15 страницAnalytic Statements Empirically Verifiable Statements: The Verification PrincipleCandy Concepcion PadizОценок пока нет
- Punctuation Marks in English GrammarДокумент3 страницыPunctuation Marks in English GrammarLesly Lisbeth Silva AcuñaОценок пока нет
- Conservation of Mass Control Volumes: By: Bashir MomoduДокумент12 страницConservation of Mass Control Volumes: By: Bashir MomoduBent ZayedОценок пока нет
- Electrical Signal and Syatem BeginnerДокумент87 страницElectrical Signal and Syatem Beginnerজীবন আহমেদОценок пока нет
- PLL PatternsДокумент6 страницPLL PatternsMax PaheinОценок пока нет
- 15 Famous Greek Mathematicians and Their ContributionsДокумент16 страниц15 Famous Greek Mathematicians and Their ContributionsMallari MarjorieОценок пока нет