Академический Документы
Профессиональный Документы
Культура Документы
(Doi 10.1021/ba-1951-0005.ch002) GREENSFELDER, B. S. - (Advances in Chemistry) PROGRESS IN PETROLEUM TECHNOLOGY Volume 5 The Mechanism of Catalytic Cracking PDF
Загружено:
Henry ArenasОригинальное название
Авторское право
Доступные форматы
Поделиться этим документом
Поделиться или встроить документ
Этот документ был вам полезен?
Это неприемлемый материал?
Пожаловаться на этот документАвторское право:
Доступные форматы
(Doi 10.1021/ba-1951-0005.ch002) GREENSFELDER, B. S. - (Advances in Chemistry) PROGRESS IN PETROLEUM TECHNOLOGY Volume 5 The Mechanism of Catalytic Cracking PDF
Загружено:
Henry ArenasАвторское право:
Доступные форматы
The Mechanism of Catalytic Cracking
B. S. GREENSFELDER
Shell Development Co., Emeryville, Calif.
The catalytic cracking of four major classes of hydro-
carbons is surveyed in terms of gas composition to
provide a basic pattern of mode of decomposition.
This pattern is correlated with the acid-catalyzed
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
low temperature reverse reactions of olefin poly-
merization and aromatic alkylation. The Whitmore
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
carbonium ion mechanism is introduced and supported
by thermochemical data, and is then applied to pro-
vide a common basis for the primary and secondary
reactions encountered in catalytic cracking and for
acid-catalyzed polymerization and alkylation re-
actions. Experimental work on the acidity of the
cracking catalyst and the nature of carbonium ions is
cited. The formation of liquid products in catalytic
cracking is reviewed briefly and the properties of the
gasoline are correlated with the over-all reaction
mechanics.
In little more than half of the 25 years covered b y this symposium, catalytic cracking
has been developed from its first acceptance to a major industrial process. I t has served
to increase the amount and octane rating of gasoline and the amounts of valuable C3 and
C4 gas components obtainable from petroleum feed stocks over those from thermal crack-
ing alone. I t is therefore of interest to seek an explanation of the nature of the products
obtained i n catalytic cracking i n terms of the hydrocarbon and catalyst chemistry which
has been developed within the past 25 years.
The first object of this paper is to set forth the basic product distributions obtained i n
the catalytic cracking of the major classes of pure hydrocarbons, which will serve to
demonstrate the action of the cracking catalyst. The second object is to assemble these
data into patterns having common denominators, to arrive at a consistent mechanism of
hydrocarbon cracking which can be specifically related to the chemical nature of the crack-
ing catalyst. T h e third is to review the experimental data available on the structural
energy relationships within and among hydrocarbons to provide real support for the pro-
posed mechanism of catalytic cracking. The proposed mechanism is then utilized to
explain a number of important secondaiy reactions encountered i n catalytic cracking
operations and to characterize the nature of catalytic gasoline to which both primary
and secondary reactions contribute.
The study of the catalytic cracking of pure hydrocarbons as a key to the interpretation
of the catalytic cracking of petroleum fractions is predicated on the belief that most of
the hydrocarbons present i n petroleum can be allocated to relatively few simple classes.
This belief is supported particularly b y the accumulated results of A P I Project 6, origi-
nally titled " T h e Separation, Identification, and Determination of the Constituents of
Petroleum" (now retitled "Analysis, Purification, and Properties of Hydrocarbons").
For the sake of consistency of experimental conditions, the data reported are those of the
3
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
4 ADVANCES IN CHEMISTRY SERIES
Shell Development C o . (5-7). F u l l acknowledgment of the work of others reported
extensively i n the literature is made thankfully, as it was of much assistance.
Accordingly, work has been done on series of -paraffins,* isoparafhns, naphthenes,
aromatics, and naphthene-aromatics which have been chosen as representative of the
major components of petroleum. I n addition, olefins, cyclo-olefins, and aromatic olefins
have been studied as a means of depicting the important secondary reactions of the copious
amounts of unsaturates produced i n the majority of catalytic cracking reactions. A
silica-zirconia-alumina catalyst was used principally; i t resembles closely i n cracking
properties typical commercial synthetic silica-alumina catalysts.
The cracking of four important classes of petroleum hydrocarbons is surveyed, using
the gaseous hydrocarbon products as a basic index of the nature of the cracking process.
Table I gives the complete gas analyses. The uniformity of cracking of n-paraffins at
500 C . may be seen from the simplified mole percentage gas compositions shown below
by carbon number on a hydrogen-free basis.
-Paraffin Ci C 2 C a C*
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
C 7 5 16 42 37
C12 9 9 42 40
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
Cie 2 6 45 47
C4 2 5 5 40 50
These gaseous products comprise from 41 to 86 weight % of the total feed reacted.
Their striking over-all consistency indicates that a uniform mode of cracking must prevail.
A very similar pattern is given b y the cracking of -olefins, which are shown next as
mole percentage gas composition. The gaseous product is now 12 to 35 weight % of the
total feed reacted for Cie and C , respectively, and is low because of the low cracking
8
temperature, 400 C. Although rarely present i n petroleum, olefins are important p r i
mary products of cracking.
-Olefin Ci C 2 C 3 C*
Ce 1 2 30 67
Cie 5 2 29 64
A t 500 C , the gaseous products from cetene and cetane become indistinguishable and
the former represents 40 weight % of the feed reacted (see Table I I ) .
The cracking at 500 C . of both monocyclic and bicyclic cyclohexane-type naphthenes,
which are important components of petroleum, again displays uniformity i n gas composi
tions, approaching that of the -paraffins. The gaseous products shown here as mole
percentage amount to 26 to 52 weight % of the total feed reacted.
Naphthene Ci C 2 C 3 C*
C 8 5 35 52
Cu 10 7 36 47
C12 4 11 37 48
Cie 9 8 36 47
I n the case of aromatics, an entirely different gas composition pattern is found. O n
selecting some of the outstanding examples of this behavior among the alkylbenzenes (6),
the striking fact emerges that the predominant gas component as mole percentage i n each
case corresponds exactly in structure to the original a l k y l group on the benzene ring.
Aromatic Ci C2 Ci n-d Iso-Ci
88 2 2
7 80 12
86
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
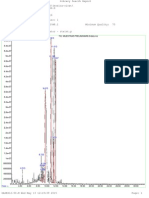
GREENSFELDERTHE MECHANISM OF CATALYTIC CRACKING 5
CARBON NUMBER
Figure 1. Catalytic Cracking Product
Distribution
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
I n a l l these examples, the total benzene plus the corresponding C or total C4 gas 3
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
components amounts to 8 9 % of the feed reacted.
Basic Cracking Patterns
F r o m these simple gas products, which correspond to a very large portion of the reacted
feed stock, two basic cracking patterns are postulated ; the first is applicable to aliphatics
and alicyclics (I) (thus including paraffins, olefins, and naphthenes), the second to sub
stituted aromatics (II). These two basic patterns are best illustrated b y Figures 1 and 2,
which show the molar distribution of the principal cracked products according to the
number of carbon atoms i n the fragments, per 100 moles of feed stock cracked, for selected
representatives of the four major hydrocarbon classes, allt 500 C. (see Table I I for
experimental conditions and product analyses).
Table I. Comparative Gas Analyses in Catalytic Cracking
(Mole % ; 1 atmosphere; silica-zirconia-alumina catalyst, Universal Oil Products Co. Type B ;
1-hour process period)
Paraffins Olefins
n-Hexadecane Parowax b n-Hexadecene
n-Heptane -Dodecane (cetane) (ca. n-tetracosane) n-Octenes (cetene)
400 C , about 7 moles per
500 C , 13.2 to 14.2 moles per liter per hour liter per hour
H, 6.5 6.5 4.9 6.1 1.7 2.2
CH 4 4.3 8.8 1.5 4.9 0.9 4.7
CH 4 8.3 5.1 3.4 2.3 0.7 1.4
CHe 6.5 3.2 2.6 2.2 1.3 1.0
CHe 25.8 25.8 32.8 30.5 26.6 23.6
CHe 14.2 13.7 10.3 7.3 2.4 4.9
I80-C4H8 6.4 8.7 10.6 11.9 17.5 11.7
n-CHe 10.6 10.4 15.8 18.4 26.5 22.7
C4H10 17.4 17.8 18.1 16.4 22.4 27.8
Run No. C-86 C-103 C-578 C-160 C-174 C-46
Naphthenes Aromatics
Isopropyl- Amyl- Triethyl- Amyl -Propyl- sec-Butyl ieri-Butyl
cyclohexane cyclohexanes cyclohexanes decalins benzene 6
benzene^ benzene**
400 and 500 C , 12.3 to 13.7
500 C , about 13 moles per liter per hour moles per liter per hour
,
CH 4
12.0
6.9n.
17.6
tm
8.2
14.4
- '
3.0 .
19.3
7.3
5.6
- >
3.5
4.4
0.0
0.6
CH 4 3.7 4.3 6.7 4.2 2.0 0.3 0!2
CHe 0.9 1.7 3.0 2.1 1.7 0.3 1.1 e
C,H e 19.5 19.5 20.7 22.1 76.2 5.4 3.5
CHe 11.1 9.9 10.7 7.2 7.4 1.1
Iso-CHe 4.8 4.4 3.8 4.8 1.0 73.4 7.3
n-CHe 8.0 8.3 8.4 9.6 1.0 3.6 76.7
CHu> 33.1 26.1 29.3 23.4 1.6/ 11.5/ 10.6*
Run No. C-146 C-156 C-145 C-144 C-150 C-542 C-540
550 C . C i - C saturates.
b
A t 6.8 moles per liter per hour. / Assume iso/n = 3.
c
500 C . Assume iso/n ~ 7
* 400 C .
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
6 ADVANCES IN CHEMISTRY SERIES
Table II. Catalytic Cracking of Representative Hydrocarbons
(500 C , 1 atmosphere; siKca-zirconia-alumina catalyst)
Amyl
Feed Cetane Cetene Decalins Cumene
Process period, min. 60 15 60 60
LHSV 4.0 7.2 3.0 1.9
Moles/liter/hour 13.6 25.0 12.8 13.7
Material balance, wt. % charge, no-loss basis
Gas 21.0 38.3 18.4 25.0
Liquid below original boiling point 17.9 56.0 51.6 54.4
Remaining liquid 60.0 3.7 28.7 16.4
Coke 1.1 2.0 1.3 4.2
Moles/100 moles cracked
Ci 4 4 11 3
C 2 16 5 9 2
C 3 112 77 42 79
C 4 116 98 55 2
C B 43 49 43
Ce 38 38 32 100
CT 7 18 18
Ce 8 9 10
Ce 7 7 12
Cio 4 6 10
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
Cu 3 4 9
Cl2 1) 8
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
Cl3 1 4 3
Cu lj 2
Cl5 2
Total 367 321 264 186
H 2 14 5 28 2
Run N o . C-578 C-1104 C-144 C-131
Carbon only.
T o obtain the first clue to the reaction mechanism, two hydrocarbons may be con
sidered: (1) 1-hexadecene (cetene), representing group I , a n d (2) isopropylbenzene
(cumene), representing group I I . W h a t common property of the catalyst will explain
the cracking patterns of both, i n conformity with what is known of the chemical reac
tions of carbon compounds?
V
CH3 CH3
6
100 90 - f 78 moles
Figure 2. Catalytic Cracking of Cumene
Based on much evidence (both from the literature and these laboratories), i t is predi
cated that the cracking reactions of cetene and cumene are directly related to the well-
known low temperature liquid or vapor phase acid-catalyzed reactions of olefin poly
merization and the alkylation of aromatics with olefins, respectively. The reciprocal
relationship of olefin polymerization and cracking is best demonstrated b y the cracking
of diisobutenes to give a gaseous product containing 73 mole % isobutene and that of
triisobutenes to give a gaseous product containing 81 mole % isobutene. T h e extension
of this reciprocal relationship of polymerization and cracking to straight-chain olefins
creates an apparent difficulty, because n-olefins polymerize to branched products. H o w
ever, the same mechanistic rules which predict the structures of such polymers also
govern the catalytic cracking of -olefins. I n the same sense, aromatic alkylation b y
olefins using acid catalysts and the cracking of a l k y l aromatics have a corresponding
reciprocal relationship. T o illustrate, the cracking of cumene gave benzene and propyl
ene, the original components, to the extent of about 86 weight % of feed cracked. I t is
concluded that the correlation between hydrocarbon cracking patterns and acidic catalysts
(both proton and "Lewis a c i d " types) is sufficient to justify further exploration of their
relationship.
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
GREENSFELDERTHE MECHANISM OF CATALYTIC CRACKING 7
The acid-catalyzed reactions of olefin polymerization and aromatic alkylation b y ole
fins have been very well explained b y the carbonium ion mechanism developed b y W h i t -
more (21). This mechanism provides the basis of the ensuing discussion, which is de
voted to the application of such concepts (7,17) to catalytic cracking systems and to the
provision of much added support i n terms of recently developed structural energy rela
tionships among hydrocarbons and new experimental evidence.
Study of the primary cracking step of the four major hydrocarbon classes leads to an
important generalization, which m a y be seen from the following type reactions :
Paraffin > paraffin + olefin
Olefin > olefin + olefin
Naphthene > saturate + olefin or olefin + olefin
Aromatic > aromatic + olefin
I n every case a n olefin is one of the products of the primary cracking step. N o w by
considering each reaction i n reverse, a common denominator for a l l the designated crack
ing systems can be found i n the chemistry of olefins. T h e answer lies i n the character
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
of the olefinic double bond, which comprises the normal valence pair electrons, and i n
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
addition two extra or " p i " electrons, which endow the double bond with the ability to
attract positively charged groups, especially protons. This ability is expressed quanti
tatively b y the "proton affinity," which is shown below for propylene and isobutylene :
AHm, K g . - C a l . / M o l e
Propylene C H = C H C H , + H+
2 > CH,CHCH, -181
+
CH, CH,
Isobutylene CH = CCH, + H
2
+
> CH,CCH, -195
+
These energy values are calculated from thermochemical tables (11) and the ionization
potentials of hydrocarl>ons obtained b y Stevenson (15) using mass spectrometric methods.
The union of an olefin and a proton from a n acid catalyst leads to the formation of a
positively charged radical, called a "carbonium i o n . " T h e two shown above are sec-
propyl and ter-butyl, respectively. [For addition to the other side of the double bond,
Ai/298 = 151.5 and 146 kg.-cal. per mole, respectively. F o r comparison, reference
is made to the older (4) values of E v a n s and Polanyi, which show differences of 7 and
21 kg.-cal. per mole between the resultant n - and s-propyl and iso-and tert-butyl ions,
respectively, against 29.5 and 49 kg.-cal. per mole here. These energy differences
control the carbonium ion isomerization reactions discussed below. ]
Such an ion may m turn combine with a second olefin :
CH 3 H
CH CH-CH + CH2=CHCH > H C i C H d C H
A *
3 3 3 3 2 3
which is the basic reaction of acid-catalyzed olefin polymerization. B y release of a
proton, the larger ion becomes the olefin polymer. T h e heats of addition of the most
important carbonium ions to a n olefinic double bond m a y be represented b y the following
figures, derived from ionization potential data of Stevenson (14,15) and thermochemical
data (11) for the reactions a l k y l ion ( R ) + ethylene, for two alternative cases:
+
R+ + H C = C H
2 2 > RCH CH + 2 2 (Reaction 1)
R+ + H C = C H
2 2 > RCHCH 3 (Reaction 2)
+
, K g . - C a l . per Mole
Ion, R + CH,+ C H 2 6
+
n-C,H 7
+
e-C,H 7
+
feri-CH 9
+
Reaction 1 -58.5 -22 -21 +7 +21.5
Reaction 2 -88 -60.5 -59.5 -31.5 -17
Reaction 3 -93 -58 -57 -29 -13.5
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
8 ADVANCES IN CHEMISTRY SERIES
Clearly, Reaction 2 is favored over Reaction 1. I n extension of Reaction 2 to higher
homologs, Reaction 3 corresponds to the union of the a l k y l ion, R , with propylene or
+
higher r*-alpha-olefins (1-CH ), to give the secondary ion, R C H 2 H ( C H ) - 3 -
2 2
CH3, with the approximate energy values listed above.
These heats of reaction also apply to the reverse reactions, which represent the crack-
ing of carbonium ions. I t is evident that on a n energy basis i t is much more difficult to
obtain methyl or ethyl ions as fragments than to obtain s-propyl or ferJ-butyl ions.
Furthermore, the release of s-propyl ion from an olefin is favored over that of n-propyl
b y about 28 kg.-cal. per mole and the release of tert-butyl ion over that of -butyl to an
even greater degree45 kg.-cal. per mole (estimated values, H). These structural
energy relationships provide the basis for the lack of Ci and C and the predominance of
2
C and C i n the gaseous products of catalytic cracking.
8 4
A l l values derived from mass spectroscopic measurements of ionization potentials are
indeed considered to be significant measures of the energy relationships among the ionic
reaction intermediates. However, further qualifications are necessary before these values
may be applied to the calculation of rates of reaction i n a specific catalytic system.
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
These qualifications are yet to be developed.
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
Formation and Reaction of the Carbonium Ion Intermediate
T o complete this picture, i t is necessary to show how the carbonium ion intermediate
is formed i n the catalytic cracking of hydrocarbons. F o r olefins, i t is the reaction of
proton addition:
H C=CH(CH )CH + H + > H C C H ( C H ) C H
2 2 3 3 2 n 3
F o r paraffins and naphthenes, the important reaction of hydride ion exchange (2) is
postulated, which is i n turn initiated b y carbonium ions derived from small amounts of
thermally produced olefins i n the cracking system.
Cracking then proceeds b y the reverse of olefin polymerization, ultimately producing
relatively noncrackable C3, C4, and C5 carbonium ions from the larger carbonium ion
intermediates. These small ions revert to olefins b y loss of a proton, which is the reverse
of the proton addition reactions illustrated previously, or become small paraffins b y the
hydride ion exchange reaction. T h e factors governing the size of the accompanying
olefinic fragment are discussed later.
Aromatics are i n a sense unique i n their catalytic cracking reactions. The aromatic
ring contains the equivalent of six double bond or p i electrons, which are, however,
mutually stabilized b y strong resonance energy. We may postulate an association be
tween a carbonium ion and these electrons i n a generalized sense:
in which the forward reaction represents the alkylation of a n aromatic and the reverse
represents the cracking of an aromatic over an acid catalyst. T h e energies of combina
tion of alkyl carbonium ions and aromatics are not known. Based on experimental
results and b y analogy with the reaction of carbonium ion and olefin, the same or similar
relative energy differences appear to govern the alkylation and cracking of aromatics.
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
GREENSFELDERTHE MECHANISM OF CATALYTIC CRACKING 9
Thus, the ease of cracking alkyi aromatics increases i n the order methyl, ethyl, isopropyl,
and ter^butylbenzene (6). This is i n exact agreement with the decreasing energies of
combination of the corresponding carbonium ions with the ethyienic double bond i n
ethylene or propylene. Confirmatory evidence for this mechanism has been obtained by
Roberts and Good b y examining the cracking of alkyl aromatics i n which the electron
density at the alkyl-aryl bond was changed in a specific manner (13).
A s portrayed above, no aromatic carbonium ion is formed as such. Rather, one
positive group is expelled as the other one enters. However, other schemes have been
suggested, such as that shown below (12,17), as well as more complicated ones which i n -
volve several resonance structures.
H
+ H +
I n a n y of these cases, a n analogy of the initiatory mechanism to that encountered
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
in olefin cracking is clear; thus, association with a proton, rather than hydride ion removal
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
(as required for paraffins and naphthenes), normally constitutes the first step i n the
cracking of both aromatics and olefins.
I n summary, the two basic types of reaction intermediates and their products are:
For paraffins, olefins, and naphthenes:
CCCR > CC=C + R +
For aromatics:
I n each case, the resultant carbonium ion, R , if large, will tend to recrack. I n
+
general, the final ion may separate into an olefin and a proton, or especially i n the cracking
of saturates, may remove a hydride ion from a neutral molecule to form a small paraffin
and a new carbonium ion. Therefore, two mechanisms are seen for the propagation of
catalytic cracking: (1) proton transfer, wherein a proton is returned to the catalyst or
donated to another molecule to regenerate the cycle; a n d (2) hydride ion exchange,
wherein a new carbonium ion is formed b y release of a hydride ion to a n existing car-
bonium ion.
A l l group I hydrocarbons (paraffins, olefins, and naphthenes) crack to give a n olefin
and a carbonium ion by the generalized mechanism:
CCCR > CC=C + R +
I t is noteworthy that the charged carbon atom of the intermediate becomes part of
the resultant olefin. The extremely important isomerization reactions of carbonium ions
determine the position of the charged atom and therefore both the size and isomeric form
of the olefinic fragment i n the primary cracking step (see 7, p. 2580, for more detailed
explanation). These isomerization reactions are governed b y the same energy rela-
tionships which enter into the proton-olefin and carbonium ion-oiefin combination energies
shown above. Thus, whenever possible, primary carbonium ions will rearrange to
secondary ions prior to cracking, so that the smallest olefin produced b y the simplest
possible type of cracking will be propylene, as shown i n the example above. Other iso-
meric, secondary ions will yield larger olefins. If rearrangement to tertiary ions takes
place prior to cracking, the smallest olefin will then be isobutylene, b y the same princ pie.
The designated mode of cracking at the carbon-carbon bond once removed from the
charged carbon atom is the simplest possible mechanism; additionally, the ionic partner,
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
10 ADVANCES IN CHEMISTRY SERIES
R , may rearrange to a secondary or tertiary ion during the cracking of the "activated
+
complex." I n reverse, these same rules successfully predict that branched-chain olefin
polymers will be obtained from either straight-chain or branched monomers. W i t h
little modification, the structures of paraffin-olefin alkylates from acid catalysts may be
predicted i n the majority of cases.
The preferential release of C and C4 as the smallest fragments is a relative matter;
3
ethylene, ethane, and methane can be produced under more drastic experimental condi-
tions, and are produced i n small amounts i n ordinary catalytic cracking. The conven-
tional process operates under conditions which maximize the desired type of splitting to
the more useful gaseous products. T o demonstrate the application of theory to practice,
the predicted and experimental curves for the cracking of cetane (7) are shown in Figure 3.
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
C A R B O N NO.
Figure 3. Catalytic Cracking of Cetane
There remains no doubt that the ionic reaction pattern of hydrocarbons is firmly
related to the presence of acid catalysts. Recent work at the Shell Development Co. (1)
and the H o u d r y Process Corp. (8) on the acid-catalyzed hydrogen-deuterium exchange
and isomerization reactions of small paraffins has brought forth strong confirmation of
the mechanistic pattern already applied to catalytic cracking. Furthermore, the work
of Thomas (17, 18), Tamele (16), and M i l l i k e n , M i l l s , and Oblad (9), among others, has
established the conventional cracking catalyst to be a n acid catalyst, capable of acting
both as a proton donor and as a strongly polar Lewis acid. T h e valences of silicon,
aluminum, and oxygen are so distributed that additional cations, such as protons, are
required for electrostatic neutrality. The physical reality of the postulated carbonium
ion intermediates is then indeed a question worthy of discussion.
Free a l k y l ions are produced under electron impact i n the high vacuum of a mass
spectrometer. They are the ions recorded b y this instrument at the cathode as an " i o n
current." I n the presence of a n acid catalyst i n a heterogeneous system containing
gaseous or liquid hydrocarbons their free existence is difficult to establish, as their nega-
tive partners must be close at hand at the surface of the catalyst. A t the San Antonio,
Tex., Southwest Regional M e e t i n g of the AMERICAN C H E M I C A L SOCIETY, December 1950,
in a Symposium on Carbonium Ions, Matsen and coworkers (S) presented cryoscopic
evidence of the existence of carbonium ions formed from 1-octene i n sulfuric acid solu-
tion. I n addition, they indicated that the carbonium ion from 1-octene was detectable
by characteristic maximum absorption i n the ultraviolet region a t 3000 to 3200 A . i n
acidic media such as sulfuric acid, phosphoric acid, and complexes such as aluminum
chloride-fer-butyl chloride and boron trifluoride-rc-propyl chloride.
The reality and the mode of existence of carbonium ions are most interesting topics
for further research i n the field of hydrocarbon chemistry.
Applications
F r o m the general principles of carbonium ion systems, a host of applications m a y be
made to important reactions of the catalytic cracking system. Some of these follow:
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
GREENSFELDERTHE MECHANISM OF CATALYTIC CRACKING 11
Preferential Saturation of Isobutylene vs. n-Butylenes
#29 Kg.-Cal./Mole (14)
c c
Ci=C+H +
> CiC -195
+
C C = C C + H+ > CCCC -186.5
+
Self-Saturation of Olefins
c c c c
C-L_cC + C C = C > C<JC + C C = C > coke (via further reactions)
+ +
Desulfurization of Mercaptans (Thiols)
CH CH CH SH + H
3 2 2
+
> CH CH CH
3 2 2 + H S>2
+
CH CH=CH + H +
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
3 2
Isomerization of Naphthenes
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
CH 3
/
C C C C
/ +
\ / X / X / \
H C
2 CH 2 H C 2 CH 2 H C
2 CH 2 H C 2 CH 2
H2 kn 2
>
H 2 i CH 2 * H C 2CH 3
/ \ / " " \ * / H, H
CH 2 CH
Double Bond Shift of Olefins
C=C-CC + H +
> CCCC > CC=CC + H+
Isomerization of Olefins
C c
c = c C C + H+ > c c c c > chc > c = c C + H+
+ +
Despite much recent progress, the energetic relationships and specific mechanistic
steps involved i n these reactions require more detailed experimental examination to pro-
vide explanation of all the observed facts and to enable more reliable prediction of new
reactions. Likewise, the specific interaction between cracking catalyst and hydro-
carbon, which also has been the subject of recent work (8, 9), is a promising field for
mechanistic studies.
Liquid Products
The liquid products of catalytic cracking (obtained i n accordance with the described
principles) have been omitted from consideration thus far, except i n the case of the a l k y l
aromatics. T o the refiner, the liquid obtained is of prime importance, both as gasoline
and heavier intermediate oils.
Paraffins produce mostly C and C liquid product, principally olefins and paraffins.
6 6
Based on feed reacted, n-Ci gave 49, n-Ci gave 44, and n-C gave 57 weight % liquid
2 6 24
product (20), under conditions given i n Table I .
Monocyclic naphthenes give relatively more cracked liquid than paraffins, primarily
because of the partial retention of rings after the cracking of side chains and because of
some dehydrogenation to aromatics. Based on feed reacted, Cu gave 59, C i gave 68, 2
and Cie gave 73 weight % liquid product (20), under conditions as i n Table I .
Bicyclic aromatics and naphthenes are important components of cracking feed stocks.
The former, after cracking i n the side chains to gasoline and gas, will remain as smaller
bicyclic aromatics in the cracked gas oil. The latter will be converted to naphthenes and
aromatics distributed i n both the gasoline and gas oil, together with aliphatic gas and
gasoline components.
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
ADVANCES IN CHEMISTRY SERIES
Olefins are usually absent from petroleum feed stocks, but they occupy a position of
great importance i n determining the character of the gasoline. I n general, the catalytic
cracking of any hydrocarbon gives at least one olefinic fragment. Such olefins are both
rapidly transformed and considerably equilibrated thermodynamically b y a number of
the ionic reactions illustrated, including double bond shift, skeletal isomerization, poly
merization, and cracking (19). F o r this reason, the gasolines produced i n the catalytic
cracking of a wide variety of petroleum feed stocks have notable similarities i n composi
tion, physical properties, and engine performance. This is i n contrast to the gasolines
obtained by thermal cracking; the absence of a catalyst capable of promoting the trans
formations of olefins makes many of the properties of thermal gasolines much more
dependent upon the composition of the feed stock and the exact conditions of cracking.
The general relationship of the cracking of pure paraffins, naphthenes, and aromatics
to that of petroleum fractions was given recently b y Voge, Good, and Greensfelder (20)
at the T h i r d World Petroleum Congress. I n general, i t was demonstrated that gasoline
yields are capable of reasonably close prediction from hydrocarbon-type analysis of the
Downloaded by CORNELL UNIV on November 24, 2012 | http://pubs.acs.org
feed stock. The presence of aromatic nitrogen bases, which specifically poison the
catalyst as shown b y M i l l s , Boedeker, and Oblad (10), makes these predictions inappli
Publication Date: January 1, 1951 | doi: 10.1021/ba-1951-0005.ch002
cable unless the nitrogen compounds are extracted; their data also provide new e v i
dence of the acidic nature of the cracking catalyst.
Summary
Fundamental studies of catalytic cracking have led to the conclusion that the chief
characteristics of the products may be traced t o the primary cracking of the hydro
carbons i n the feed stock and to the secondary reactions of the olefins produced; both
correspond to the ionic reaction mechanisms of hydrocarbons i n the presence of acidic
catalysts. The chemistry of both the hydrocarbons and catalysts dealt with here has
advanced rapidly i n the last decade. Nevertheless, much further exploration is required
with respect to the nature of the catalyst and the properties of the hydrocarbons under
going reaction. A promising field lies ahead for future research.
Acknowledgment
The assistance of G . M . Good, D . P . Stevenson, and H . H . Voge, and of M r s . A .
Carruth of the Shell Development C o . i n the preparation of this paper is gratefully
acknowledged.
Literature Cited
(1) Beeck, O., Otvos, J . W., Stevenson, D. P., and Wagner, C . D., J. Chem. Phys., 16, 255 (1948);
17, 418, 419 (1949).
(2) Brewer, C . P., and Greensfelder, B. S., J. Am. Chem.Soc.,73, 2257 (1951).
(3) Chem. Eng. News, 28, 4552 (1950).
(4) Evans, A. G., and Polanyi, M., J. Chem.Soc.,1947, 252.
(5) Greensfelder, B. S., and Voge, H . H., Ind. Eng. Chem., 37, 514, 983, 1038 (1945).
(6) Greensfelder, B. S., Voge, H . H., and Good, G . M., Ibid., 37, 1168 (1945).
(7) Ibid., 41, 2573 (1949).
(8) Hindin, S. G., Mills, G. ., and Oblad, A. G., J. Am. Chem.Soc.,73, 278 (1951).
(9) Milliken, T. H., Jr., Mills, G. ., and Oblad, A . G., Faraday Soc. Discussions, 8, 279 (1950).
(10) Mills, G. ., Boedeker, E . R., and Oblad, A . G., J. Am. Chem.Soc.,72, 1554 (1950).
(11) Natl. Bur. Standards, Circ C-461 (1950).
(12) Price, C . C., Chem. Revs., 29, 37 (1941).
(13) Roberts, R. M., and Good, G . M., J. Am. Chem.Soc.,73, 1320 (1951).
(14) Shell Development Co., unpublished work.
(15) Stevenson, D. P., Faraday Society Discussion on Hydrocarbons, April 1951.
(16) Tamele, M . W., FaradaySoc.Discussions, 8, 270 (1950).
(17) Thomas, C. L., Ind. Eng. Chem., 41, 2564 (1949).
(18) Thomas, C. L., Hickey, J . , and Stecker, G., Ibid., 42, 866 (1950).
(19) Voge, . H., Good, G . M., and Greensfelder, B. S., Ibid., 38, 1033 (1946).
(20) Voge, . H., Good, G . M., and Greensfelder, B. S., Proc. Third World Petroleum Congress,
The Hague, 1951.
(21) Whitmore, F. C., Chem. Eng. News, 26, 668 (1948).
R E C E I V E D M a y 16, 1951.
In PROGRESS IN PETROLEUM TECHNOLOGY;
Advances in Chemistry; American Chemical Society: Washington, DC, 1951.
Вам также может понравиться
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (119)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2219)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- (9781585286010 - Basic Concepts in Medicinal Chemistry) Answers To Chapter QuestionsДокумент116 страниц(9781585286010 - Basic Concepts in Medicinal Chemistry) Answers To Chapter QuestionsShrinivas JahagirdarОценок пока нет
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- Nitration of Methyl BenzoateДокумент3 страницыNitration of Methyl BenzoateDaniel McDermottОценок пока нет
- Design and Development of Downdraft Gasifier For Operating CI Engine On Dual Fuel Mode PDFДокумент78 страницDesign and Development of Downdraft Gasifier For Operating CI Engine On Dual Fuel Mode PDFHenry ArenasОценок пока нет
- The Following Are Some of The Most Difficult Questions You Will Face in The CourseДокумент27 страницThe Following Are Some of The Most Difficult Questions You Will Face in The CourseHenry ArenasОценок пока нет
- Crecer Haciendo Que Otros Crezcan Es Una de Las Premisas Que Motivan Nuestro Trabajo en Pan American Energy Desde Hace 20 Años", Explicó Huergo"Документ3 страницыCrecer Haciendo Que Otros Crezcan Es Una de Las Premisas Que Motivan Nuestro Trabajo en Pan American Energy Desde Hace 20 Años", Explicó Huergo"Henry ArenasОценок пока нет
- Dermatan Sulfate - Chitosan Polyelectrolyte Complex With Potential Application in The Treatment and Diagnosis of Vascular DiseaseДокумент9 страницDermatan Sulfate - Chitosan Polyelectrolyte Complex With Potential Application in The Treatment and Diagnosis of Vascular DiseaseHenry ArenasОценок пока нет
- Cinetica PsicrofilicaДокумент11 страницCinetica PsicrofilicaHenry ArenasОценок пока нет
- Gasifier ProcessДокумент4 страницыGasifier ProcessHenry ArenasОценок пока нет
- WeightedДокумент28 страницWeightedHenry ArenasОценок пока нет
- MN TLC CatalogueДокумент28 страницMN TLC CatalogueHenry ArenasОценок пока нет
- 1961 - Symposium (International) On Combustion - The Activation Energy of Gas - TesnerДокумент8 страниц1961 - Symposium (International) On Combustion - The Activation Energy of Gas - TesnerHenry ArenasОценок пока нет
- 1 s2.0 S0306261914008307 MainДокумент7 страниц1 s2.0 S0306261914008307 MainHenry ArenasОценок пока нет
- New Drive For Chinas Coal Industry - Reference PDFДокумент12 страницNew Drive For Chinas Coal Industry - Reference PDFHenry ArenasОценок пока нет
- Articulo Evol de Bcterias PDFДокумент4 страницыArticulo Evol de Bcterias PDFHenry ArenasОценок пока нет
- Development of the Shell-Koppers Coal Gasification ProcessДокумент11 страницDevelopment of the Shell-Koppers Coal Gasification Processhen1911Оценок пока нет
- 渣浆泵 选型参数表Operating Conditions (Eng-Chn)Документ1 страница渣浆泵 选型参数表Operating Conditions (Eng-Chn)hen1911Оценок пока нет
- 1961 - Symposium (International) On Combustion - The Activation Energy of Gas - TesnerДокумент8 страниц1961 - Symposium (International) On Combustion - The Activation Energy of Gas - TesnerHenry ArenasОценок пока нет
- (Doi 10.1002/0471238961), - Kirk-Othmer Encyclopedia of Chemical TechnologyДокумент13 страниц(Doi 10.1002/0471238961), - Kirk-Othmer Encyclopedia of Chemical TechnologyHenry ArenasОценок пока нет
- 1961 - Symposium (International) On Combustion - The Activation Energy of Gas - TesnerДокумент8 страниц1961 - Symposium (International) On Combustion - The Activation Energy of Gas - TesnerHenry ArenasОценок пока нет
- Internal-Combustion Engine in Theory and Practice, Volume 2 - Combustion, Fuels, Materials, Design (2nd Edition, Revised) © 1985 MIT Press 1Документ2 страницыInternal-Combustion Engine in Theory and Practice, Volume 2 - Combustion, Fuels, Materials, Design (2nd Edition, Revised) © 1985 MIT Press 1Henry ArenasОценок пока нет
- 1 s2.0 S0960852412018512 MainДокумент7 страниц1 s2.0 S0960852412018512 MainHenry ArenasОценок пока нет
- (Doi 10.1002/0471238961), - Kirk-Othmer Encyclopedia of Chemical TechnologyДокумент13 страниц(Doi 10.1002/0471238961), - Kirk-Othmer Encyclopedia of Chemical TechnologyHenry ArenasОценок пока нет
- Basic Design of a Fluidized Bed Gasifier for Rice HuskДокумент8 страницBasic Design of a Fluidized Bed Gasifier for Rice HuskAnna Yunita SitompulОценок пока нет
- GasifierДокумент192 страницыGasifierHenry ArenasОценок пока нет
- Catalysis in The Refining of Fischer-Tropsch Syncrude © 2010 Royal Society of Chemistry 1Документ2 страницыCatalysis in The Refining of Fischer-Tropsch Syncrude © 2010 Royal Society of Chemistry 1Henry ArenasОценок пока нет
- Material Lower Flammability Limit (Vol%) Upper Flammability Limit (Vol%) Petrol (Gasoline) 1.3 7Документ1 страницаMaterial Lower Flammability Limit (Vol%) Upper Flammability Limit (Vol%) Petrol (Gasoline) 1.3 7Henry ArenasОценок пока нет
- GerixДокумент1 страницаGerixHenry ArenasОценок пока нет
- Qualmuestra5 Preliminar6.dДокумент12 страницQualmuestra5 Preliminar6.dHenry ArenasОценок пока нет
- Calibration of Volumetric GlasswareДокумент6 страницCalibration of Volumetric GlasswareHenry ArenasОценок пока нет
- Articulo ProyectoДокумент9 страницArticulo ProyectoHenry ArenasОценок пока нет
- Articulo ProyectoДокумент9 страницArticulo ProyectoHenry ArenasОценок пока нет
- 13 Goc Revision Notes QuizrrДокумент146 страниц13 Goc Revision Notes QuizrrDHRUV WORLDОценок пока нет
- R315 AbenojaJL HYDROCARBONSДокумент8 страницR315 AbenojaJL HYDROCARBONSJL AbenojaОценок пока нет
- Chemistry 120.1 - Organic Chemistry Laboratory Laboratory ReportДокумент3 страницыChemistry 120.1 - Organic Chemistry Laboratory Laboratory Reportkat katОценок пока нет
- Aromatic CompoundsДокумент6 страницAromatic CompoundsSANIA FAJAR KHANОценок пока нет
- ICT Mumbai B.Chem - Engg SyllabusДокумент41 страницаICT Mumbai B.Chem - Engg SyllabusAniruddha ShidhayeОценок пока нет
- Organic Halide Classification TestsДокумент8 страницOrganic Halide Classification Testsjullian marasiganОценок пока нет
- Organic Chemistry Mechanistic Patterns Canadian 1st Edition Ogilvie Solutions ManualДокумент23 страницыOrganic Chemistry Mechanistic Patterns Canadian 1st Edition Ogilvie Solutions Manualslapperboatbill49x100% (27)
- Understanding Isocyanate Chemistry and ReactivityДокумент15 страницUnderstanding Isocyanate Chemistry and Reactivitymita shilОценок пока нет
- Resonance and Induction Tutorial: Jack DeruiterДокумент19 страницResonance and Induction Tutorial: Jack DeruiterDanish AhmedОценок пока нет
- Hydrocarbons Class 11 Notes Chemistry Chapter 13 - Learn CBSEДокумент16 страницHydrocarbons Class 11 Notes Chemistry Chapter 13 - Learn CBSERishabh Singh RajputОценок пока нет
- 7 B Kof Homo Lumo PDFДокумент27 страниц7 B Kof Homo Lumo PDFIstiОценок пока нет
- Heterocycles, Their Synthesis and Industrial Applications: A ReviewДокумент22 страницыHeterocycles, Their Synthesis and Industrial Applications: A ReviewIJRASETPublicationsОценок пока нет
- COKE FORMATION IN CATALYTIC CRACKINGДокумент9 страницCOKE FORMATION IN CATALYTIC CRACKINGwiboonwiОценок пока нет
- Hydrocarbon PDFДокумент19 страницHydrocarbon PDFPrincess Lou CarpenteroОценок пока нет
- Himanshu Pandey SolutionsДокумент144 страницыHimanshu Pandey SolutionsDeepak S.V.73% (48)
- SYLLABUS FOR ENTRANCE EXAMДокумент11 страницSYLLABUS FOR ENTRANCE EXAMMahesh ShahОценок пока нет
- 23 AminesДокумент52 страницы23 AminesGoka Agbesi GokaОценок пока нет
- Nitration of Methyl Benzoate: Vacuum Filtration and RecrystallizationДокумент23 страницыNitration of Methyl Benzoate: Vacuum Filtration and RecrystallizationgudupopsОценок пока нет
- Wellhead Corrosion and Trim SelectionДокумент34 страницыWellhead Corrosion and Trim SelectionGuillaume Boyer100% (3)
- Test Bank For General Organic and Biochemistry 8th Edition Katherine Denniston Download Full DownloadДокумент19 страницTest Bank For General Organic and Biochemistry 8th Edition Katherine Denniston Download Full Downloadmarcjohnstontsbgmqofip100% (35)
- GATE-PYQ-Topicwise Noor Ul Huda SirДокумент585 страницGATE-PYQ-Topicwise Noor Ul Huda SirSudha balakrishnan100% (2)
- Solomons ch14 MedicineДокумент44 страницыSolomons ch14 MedicineAhmad GasheemОценок пока нет
- History of benzene ring discovery by Kekule in 1865Документ1 страницаHistory of benzene ring discovery by Kekule in 1865ekadarma55100% (5)
- Aromatic CompoundДокумент256 страницAromatic CompoundLuc LeОценок пока нет
- EXPE9Документ8 страницEXPE9K-yanVehraaYomomaОценок пока нет
- Petrochemical Industry - Production ProcessДокумент40 страницPetrochemical Industry - Production ProcessAyie Arie AyitОценок пока нет
- Molecular structural studies of key lichen compoundsДокумент11 страницMolecular structural studies of key lichen compoundsArif GunawanОценок пока нет