Вам также может понравиться
- Final Exam Review: Appointments Will Be Happening Monday Afternoon and Before Final On WednesdayДокумент91 страницаFinal Exam Review: Appointments Will Be Happening Monday Afternoon and Before Final On WednesdayDonny Jinjae Kim-adamsОценок пока нет
- Lec4 - Multiple Sequence AlignmentДокумент22 страницыLec4 - Multiple Sequence AlignmentmbathulaОценок пока нет
- C SharpДокумент144 страницыC SharpAlisha SinghalОценок пока нет
- Lesson 3Документ54 страницыLesson 3Ken_nerveОценок пока нет
- UNEX GIS WSAnalysis AdvArc10 Python PDFДокумент34 страницыUNEX GIS WSAnalysis AdvArc10 Python PDFgokonkОценок пока нет
- Database Similarity Searching: Irit Orr Shifra Ben DorДокумент76 страницDatabase Similarity Searching: Irit Orr Shifra Ben DorRishabh JainОценок пока нет
- Chap 2 Pt. 2 String ClassДокумент24 страницыChap 2 Pt. 2 String ClasspjsssssoОценок пока нет
- Mid s18 SolДокумент11 страницMid s18 SolRaJu SinGhОценок пока нет
- Lecture2022 - 3 /!Документ60 страницLecture2022 - 3 /!MAnugrahRizkyPОценок пока нет
- Lesson 02 - Built-In ClassesДокумент34 страницыLesson 02 - Built-In ClassesБогдан КотикОценок пока нет
- 23 JavascriptДокумент28 страниц23 JavascriptMazharОценок пока нет
- Crash Course in Java: - Network Programming in Java Is Very Different Than in C/C++Документ10 страницCrash Course in Java: - Network Programming in Java Is Very Different Than in C/C++Neville NiuОценок пока нет
- String 1.1Документ34 страницыString 1.1Akash SavaliyaОценок пока нет
- Chapter 9Документ96 страницChapter 9memer babaОценок пока нет
- PythonДокумент35 страницPythonsahardevjani635Оценок пока нет
- Lecture 1 - Abstraction and ListsДокумент14 страницLecture 1 - Abstraction and ListsrasОценок пока нет
- Pig HiveДокумент58 страницPig HivePriyanki TanwarОценок пока нет
- Week13 14 Arrays Part2 StudentДокумент49 страницWeek13 14 Arrays Part2 Studentjiwonhyung0234Оценок пока нет
- JAva InterviewДокумент13 страницJAva InterviewpnitinmОценок пока нет
- 1.1 Algorithm ParadigmsДокумент32 страницы1.1 Algorithm ParadigmsStanley WangОценок пока нет
- Lab MannualДокумент49 страницLab Mannualvickyakfan152002Оценок пока нет
- Finding Bugs in Database Systems Via Query Partitioning: Conference PaperДокумент31 страницаFinding Bugs in Database Systems Via Query Partitioning: Conference PaperYogita SoniОценок пока нет
- Concurrent Programiing Tutorial-2Документ8 страницConcurrent Programiing Tutorial-2niku007Оценок пока нет
- The ORCA Quantum Chemistry Program: A Jump-Start GuideДокумент6 страницThe ORCA Quantum Chemistry Program: A Jump-Start GuideFerzz MontejoОценок пока нет
- Random ForestДокумент83 страницыRandom ForestBharath Reddy MannemОценок пока нет
- Lecture Open MPДокумент25 страницLecture Open MPDr. V. Padmavathi Associate ProfessorОценок пока нет
- CSE2003 Data-Structures-And-Algorithms ETH 1 AC39Документ11 страницCSE2003 Data-Structures-And-Algorithms ETH 1 AC39heenaОценок пока нет
- Basic PythonДокумент40 страницBasic Python20021382 Phạm Công LânОценок пока нет
- Datalog: Deductive Database Programming: Jay Mccarthy Jay@Racket-LangДокумент13 страницDatalog: Deductive Database Programming: Jay Mccarthy Jay@Racket-LangJuanjo GGzzОценок пока нет
- Data Structures and AlgorithmsДокумент115 страницData Structures and Algorithmsjames oshomahОценок пока нет
- AI Lab Manual 1Документ13 страницAI Lab Manual 1Mariyam AliОценок пока нет
- Buffer Overflow Exploits: Taken Shamelessly From: /courses/cse451/05sp/section/ove Rflow1Документ27 страницBuffer Overflow Exploits: Taken Shamelessly From: /courses/cse451/05sp/section/ove Rflow1Avinash AvuthuОценок пока нет
- DR.C S Prasanth-PhysicsДокумент42 страницыDR.C S Prasanth-PhysicsAsian DeckersОценок пока нет
- Chapter9 Part2Документ16 страницChapter9 Part2Artemis ZeusbornОценок пока нет
- Lecture 22.1-Python DatatypesДокумент43 страницыLecture 22.1-Python DatatypesDileep LОценок пока нет
- CH-6 More About Classes & Libraries PDFДокумент3 страницыCH-6 More About Classes & Libraries PDFJay SandukeОценок пока нет
- 10 Caches DetailДокумент35 страниц10 Caches DetailSudhir SainiОценок пока нет
- Unit 5 - Runtime EnvironmentДокумент69 страницUnit 5 - Runtime Environmentaxar kumarОценок пока нет
- Dynamic Array Data Structure - Interview CakeДокумент9 страницDynamic Array Data Structure - Interview CakeRonaldMartinezОценок пока нет
- Lecture 11Документ41 страницаLecture 11myturtle game01Оценок пока нет
- 4 DataAnalyics Part1Документ59 страниц4 DataAnalyics Part1Ali Shana'aОценок пока нет
- BDA02 IntroToPythonДокумент34 страницыBDA02 IntroToPythonGargi Jana100% (1)
- OS Intro To Scientific ComputingДокумент35 страницOS Intro To Scientific ComputingMauricio Nahún González GuevaraОценок пока нет
- Lec 02-04 Ch2 Numpy Part 1Документ66 страницLec 02-04 Ch2 Numpy Part 1MAryam KhanОценок пока нет
- 02 BasicsДокумент35 страниц02 BasicsDaudet Polycarpe TiomelaОценок пока нет
- DSA & Java LecturesДокумент54 страницыDSA & Java Lectures235KEVIN CHANGANIОценок пока нет
- 6.1 Overflow1Документ27 страниц6.1 Overflow1coeus koalemosОценок пока нет
- Mid s18Документ12 страницMid s18RaJu SinGhОценок пока нет
- JavaScript Data Structures and Algorithms: An Introduction to Understanding and Implementing Core Data Structure and Algorithm FundamentalsОт EverandJavaScript Data Structures and Algorithms: An Introduction to Understanding and Implementing Core Data Structure and Algorithm FundamentalsОценок пока нет
- cs224n Python Review 20Документ47 страницcs224n Python Review 20JillОценок пока нет
- Lect 5Документ43 страницыLect 5Ananya JainОценок пока нет
- Python For Data Science Nympy and PandasДокумент4 страницыPython For Data Science Nympy and PandasStocknEarnОценок пока нет
- Django ModelsДокумент86 страницDjango ModelsCF TiaretОценок пока нет
- An Introduction To Answer Set Programming: Paul Vicol October 14, 2015 (Based On Slides by Torsten Schaub)Документ51 страницаAn Introduction To Answer Set Programming: Paul Vicol October 14, 2015 (Based On Slides by Torsten Schaub)Warley GramachoОценок пока нет
- CS3362 - Data Science Laboratory - Manual - Final-1Документ76 страницCS3362 - Data Science Laboratory - Manual - Final-1Ramapriya. K 21CSA69Оценок пока нет
- Data Structure&GenericsДокумент60 страницData Structure&GenericssanalОценок пока нет
- Java IntroДокумент41 страницаJava IntroVikram BhuskuteОценок пока нет
- Building Java Programs: Linked ListsДокумент97 страницBuilding Java Programs: Linked ListsMinh NguyenОценок пока нет
- Architecting Petabyte-Scale Analytics by Scaling Out Postgres On Azure With CitusДокумент32 страницыArchitecting Petabyte-Scale Analytics by Scaling Out Postgres On Azure With CitusOzioma IhekwoabaОценок пока нет
- Better APIs Faster Tests and More Resilient Systems With Spring Cloud Contract 1Документ79 страницBetter APIs Faster Tests and More Resilient Systems With Spring Cloud Contract 1Ozioma IhekwoabaОценок пока нет
- Integrating PM SM PDFДокумент15 страницIntegrating PM SM PDFOzioma IhekwoabaОценок пока нет
- What's New in Spring Cloud?: Oleg Zhurakousky Olga Maciaszek-SharmaДокумент36 страницWhat's New in Spring Cloud?: Oleg Zhurakousky Olga Maciaszek-SharmaOzioma IhekwoabaОценок пока нет
- Sample SLA Templates PDFДокумент14 страницSample SLA Templates PDFOzioma Ihekwoaba100% (2)
- Accelerating Electronic Trading: Mellanox Adapter CardsДокумент2 страницыAccelerating Electronic Trading: Mellanox Adapter CardsOzioma IhekwoabaОценок пока нет
- 2012 Solarflare Openonload Performance Brief 10Документ11 страниц2012 Solarflare Openonload Performance Brief 10Ozioma IhekwoabaОценок пока нет
- Survival Data Mining For Customer Insight: Prepared By: Gordon S. Linoff Data Miners June 2004Документ10 страницSurvival Data Mining For Customer Insight: Prepared By: Gordon S. Linoff Data Miners June 2004awesomelycrazyОценок пока нет
- MobiusДокумент10 страницMobiusOzioma IhekwoabaОценок пока нет
- Vignesh Sukumar SVCC 2012Документ25 страницVignesh Sukumar SVCC 2012Ozioma IhekwoabaОценок пока нет
- Part3 BinaryChoice InferenceДокумент71 страницаPart3 BinaryChoice InferenceOzioma IhekwoabaОценок пока нет
- Flink Batch BasicsДокумент37 страницFlink Batch BasicsOzioma IhekwoabaОценок пока нет
- Apache Big Data Europe Dashdb Final2Документ19 страницApache Big Data Europe Dashdb Final2Ozioma IhekwoabaОценок пока нет
- Real Time Analytics With Apache Kafka and Spark: @rahuldausaДокумент54 страницыReal Time Analytics With Apache Kafka and Spark: @rahuldausaOzioma IhekwoabaОценок пока нет
- Defining The Big Data Architecture FrameworkДокумент55 страницDefining The Big Data Architecture FrameworkOzioma IhekwoabaОценок пока нет
- Reactivestreams 140507092422 Phpapp01Документ60 страницReactivestreams 140507092422 Phpapp01Andres Tuells JanssonОценок пока нет
- Big Data Media 2398958Документ27 страницBig Data Media 2398958Ozioma IhekwoabaОценок пока нет
- Big Data Oil Gas 2515144 PDFДокумент24 страницыBig Data Oil Gas 2515144 PDFTim ClarkeОценок пока нет
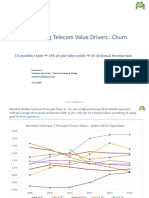
- Churn Prediction Model HildaДокумент49 страницChurn Prediction Model HildaOzioma IhekwoabaОценок пока нет
- Improving Manufacturing Performance With Big Data: Architect's Guide and Reference Architecture IntroductionДокумент23 страницыImproving Manufacturing Performance With Big Data: Architect's Guide and Reference Architecture IntroductionOzioma IhekwoabaОценок пока нет
- Big Data Utilities 2398956Документ25 страницBig Data Utilities 2398956Ozioma IhekwoabaОценок пока нет
- CDR Sample Data ModelДокумент24 страницыCDR Sample Data ModelMani TiwaryОценок пока нет
- Big Data Retail 2398957Документ25 страницBig Data Retail 2398957Ozioma IhekwoabaОценок пока нет
- Paper 4-Churn Prediction in Telecommunication PDFДокумент3 страницыPaper 4-Churn Prediction in Telecommunication PDFOzioma IhekwoabaОценок пока нет
- 1monthlychurnequals24ofsubsareswitching 141002064344 Phpapp01Документ7 страниц1monthlychurnequals24ofsubsareswitching 141002064344 Phpapp01Ozioma IhekwoabaОценок пока нет
- Hand Book On Bccs in GSM Ks Krishnan Sdetrichy1Документ19 страницHand Book On Bccs in GSM Ks Krishnan Sdetrichy1dkprakash113Оценок пока нет
- A Case Study Report On Churn Analysis: Submitted To Mr. Sanjay Rao Founder, Axion Connect Presented by Amit KumarДокумент8 страницA Case Study Report On Churn Analysis: Submitted To Mr. Sanjay Rao Founder, Axion Connect Presented by Amit KumarOzioma IhekwoabaОценок пока нет
- Churn Modeling For Mobile Telecommunicationsnewptt 120220124603 Phpapp02Документ21 страницаChurn Modeling For Mobile Telecommunicationsnewptt 120220124603 Phpapp02Ozioma IhekwoabaОценок пока нет
- Telecoms Billing ManualДокумент27 страницTelecoms Billing ManualKhalid Makahleh100% (1)
- 09a.billing SystemsДокумент26 страниц09a.billing SystemsNeeraj SahuОценок пока нет
- Case 3 SectionC Group 1 (Repaired)Документ3 страницыCase 3 SectionC Group 1 (Repaired)SANDEEP AGRAWALОценок пока нет
- Bench-Scale Decomposition of Aluminum Chloride Hexahydrate To Produce Poly (Aluminum Chloride)Документ5 страницBench-Scale Decomposition of Aluminum Chloride Hexahydrate To Produce Poly (Aluminum Chloride)varadjoshi41Оценок пока нет
- DNA Vs RNA - Introduction and Differences Between DNA and RNAДокумент10 страницDNA Vs RNA - Introduction and Differences Between DNA and RNAKienlevyОценок пока нет
- UFO Yukon Spring 2010Документ8 страницUFO Yukon Spring 2010Joy SimsОценок пока нет
- C. Robert Mesle (Auth.) - John Hick's Theodicy - A Process Humanist Critique-Palgrave Macmillan UK (1991)Документ168 страницC. Robert Mesle (Auth.) - John Hick's Theodicy - A Process Humanist Critique-Palgrave Macmillan UK (1991)Nelson100% (3)
- MDI - Good Fellas - ScriptДокумент20 страницMDI - Good Fellas - ScriptRahulSamaddarОценок пока нет
- Bioinformatics Computing II: MotivationДокумент7 страницBioinformatics Computing II: MotivationTasmia SaleemОценок пока нет
- Natural Cataclysms and Global ProblemsДокумент622 страницыNatural Cataclysms and Global ProblemsphphdОценок пока нет
- Vocabulary FceДокумент17 страницVocabulary Fceivaan94Оценок пока нет
- B. Inggris X - 7Документ8 страницB. Inggris X - 7KabardiantoОценок пока нет
- Contemporary Philippine Arts From The Regions: Quarter 1Документ11 страницContemporary Philippine Arts From The Regions: Quarter 1JUN GERONAОценок пока нет
- Group 2 ITI Consensus Report: Prosthodontics and Implant DentistryДокумент9 страницGroup 2 ITI Consensus Report: Prosthodontics and Implant DentistryEsme ValenciaОценок пока нет
- FINAL BÁO-CÁO-THỰC-TẬP.editedДокумент38 страницFINAL BÁO-CÁO-THỰC-TẬP.editedngocthaongothi4Оценок пока нет
- Erosional VelocityДокумент15 страницErosional VelocityGary JonesОценок пока нет
- EngHub How To Break HabitsДокумент13 страницEngHub How To Break HabitsViktoria NovikovaОценок пока нет
- Action ResearchДокумент2 страницыAction ResearchGeli BaringОценок пока нет
- Dessler HRM12e PPT 01Документ30 страницDessler HRM12e PPT 01harryjohnlyallОценок пока нет
- Chapter 5 - CheerdanceДокумент10 страницChapter 5 - CheerdanceJoana CampoОценок пока нет
- Java Complete Collection FrameworkДокумент28 страницJava Complete Collection FrameworkkhushivanshОценок пока нет
- Introduction To Password Cracking Part 1Документ8 страницIntroduction To Password Cracking Part 1Tibyan MuhammedОценок пока нет
- Working Capital Management 2012 of HINDALCO INDUSTRIES LTD.Документ98 страницWorking Capital Management 2012 of HINDALCO INDUSTRIES LTD.Pratyush Dubey100% (1)
- Cipet Bhubaneswar Skill Development CoursesДокумент1 страницаCipet Bhubaneswar Skill Development CoursesDivakar PanigrahiОценок пока нет
- SAFE RC Design ForДокумент425 страницSAFE RC Design ForMarlon Braggian Burgos FloresОценок пока нет
- Rishika Reddy Art Integrated ActivityДокумент11 страницRishika Reddy Art Integrated ActivityRishika ReddyОценок пока нет
- Cpar Characteristics and Functions Week 3Документ128 страницCpar Characteristics and Functions Week 3christianwood0117Оценок пока нет
- Gujarat Urja Vikas Nigam LTD., Vadodara: Request For ProposalДокумент18 страницGujarat Urja Vikas Nigam LTD., Vadodara: Request For ProposalABCDОценок пока нет
- Priest, Graham - The Logic of The Catuskoti (2010)Документ31 страницаPriest, Graham - The Logic of The Catuskoti (2010)Alan Ruiz100% (1)
- Electrical Engineering Lab Vica AnДокумент6 страницElectrical Engineering Lab Vica Anabdulnaveed50% (2)
- Project Chalk CorrectionДокумент85 страницProject Chalk CorrectionEmeka Nicholas Ibekwe100% (6)
- Marine Cargo InsuranceДокумент72 страницыMarine Cargo InsuranceKhanh Duyen Nguyen HuynhОценок пока нет