Вам также может понравиться
- Drenajes GeomorfologiaДокумент14 страницDrenajes GeomorfologiaJorge L. Gutierrez B.Оценок пока нет
- Karnawati - Mekanisme Gerakan Massa BatuanДокумент13 страницKarnawati - Mekanisme Gerakan Massa BatuanTri SamosirОценок пока нет
- Rundown Minggu 14 April 2019Документ1 страницаRundown Minggu 14 April 2019Nia LalombombuidaОценок пока нет
- DafpusДокумент1 страницаDafpusNia LalombombuidaОценок пока нет
- Lion MitaДокумент1 страницаLion MitaNia LalombombuidaОценок пока нет
- 1Документ1 страница1Nia LalombombuidaОценок пока нет
- KELOMPOK 1 TANGGAL 2/9/2017 Koordinat AZIMUTH N 15°E LOKASI Bantul SPASI 5m FREKUENSI 22300 PANJANG 115 m LINTASAN OPERATOR TogetherДокумент28 страницKELOMPOK 1 TANGGAL 2/9/2017 Koordinat AZIMUTH N 15°E LOKASI Bantul SPASI 5m FREKUENSI 22300 PANJANG 115 m LINTASAN OPERATOR TogetherNia LalombombuidaОценок пока нет
- TekKom 10 Multimedia and The Web PDFДокумент13 страницTekKom 10 Multimedia and The Web PDFNia LalombombuidaОценок пока нет
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5784)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (890)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (119)
- Liver Mori UmДокумент3 страницыLiver Mori UmKevinОценок пока нет
- Varsity Science Questions - ChemistryДокумент20 страницVarsity Science Questions - ChemistrySlasher124Оценок пока нет
- SALTДокумент22 страницыSALTparitoshОценок пока нет
- 4CH1 1C Que 20210428Документ28 страниц4CH1 1C Que 20210428Yağmur AtunОценок пока нет
- Arsenic, Antimony and Bismuth: Stibium AntimoniumДокумент7 страницArsenic, Antimony and Bismuth: Stibium AntimoniumlaythОценок пока нет
- Steel GradesДокумент1 страницаSteel GradesBoris TrostyanezkyОценок пока нет
- DS-4, English MediumДокумент56 страницDS-4, English MediumRashini AnneОценок пока нет
- Chemical EquationsДокумент10 страницChemical Equationsme-elormОценок пока нет
- Properties of A 55 % Aluminium Zinc Coating On Steel Sheeting PDFДокумент4 страницыProperties of A 55 % Aluminium Zinc Coating On Steel Sheeting PDFing_fernandogalvez2015Оценок пока нет
- Welding For Nickel AlloysДокумент20 страницWelding For Nickel AlloysClodomiro Barril Club de Amigos de la CuecaОценок пока нет
- Born-Haber cycle for NaClДокумент458 страницBorn-Haber cycle for NaCladalinefallingstar100% (1)
- 1chemical Reactions & Equations Top 25 Questions Prashant KiradДокумент12 страниц1chemical Reactions & Equations Top 25 Questions Prashant KiradKshitiz sharma100% (1)
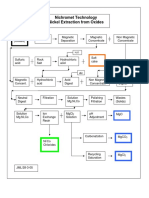
- Nickel Extraction DiagramsДокумент2 страницыNickel Extraction DiagramsDan MatОценок пока нет
- Analisa Pertambangan Timah Di Provinsi Kepulauan Bangka BelitungДокумент9 страницAnalisa Pertambangan Timah Di Provinsi Kepulauan Bangka BelitungBima Maulana PutraОценок пока нет
- SRI CHAITANYA IIT ACADEMY CHEMISTRY DOCUMENTДокумент19 страницSRI CHAITANYA IIT ACADEMY CHEMISTRY DOCUMENTggk20130% (2)
- Chapter 19-Oxidation-Reduction ReactionsДокумент22 страницыChapter 19-Oxidation-Reduction ReactionsNada MeselhyОценок пока нет
- Steel NumbersДокумент3 страницыSteel NumbersAhmadiBinAhmadОценок пока нет
- ChromiumДокумент2 страницыChromiumKaty XiomaraОценок пока нет
- AGAT, Bureau VeritasДокумент30 страницAGAT, Bureau Veritasgavrilenko1426Оценок пока нет
- Topical preparations and inorganic gases used in pharmaceuticalsДокумент5 страницTopical preparations and inorganic gases used in pharmaceuticalssubeesh up100% (1)
- A29A29M-12e1 Standard Specification For General Requirements For Steel Bars, Carbon and Alloy, Hot-Wrought 000Документ16 страницA29A29M-12e1 Standard Specification For General Requirements For Steel Bars, Carbon and Alloy, Hot-Wrought 000Moongil oodagamОценок пока нет
- Topic18 AnswersДокумент16 страницTopic18 AnswersHeaven WincletОценок пока нет
- S Stage 8 P110 01 AFP PDFДокумент18 страницS Stage 8 P110 01 AFP PDFPunitha PanchaОценок пока нет
- ASTM Coal Testing Standards ListДокумент2 страницыASTM Coal Testing Standards ListYantiyanti IIОценок пока нет
- 02-Stoichiometry WS 1Документ2 страницы02-Stoichiometry WS 1rhaineОценок пока нет
- Chemical Calculations 1Документ10 страницChemical Calculations 1Garcia RaphОценок пока нет
- Redox TitrationДокумент4 страницыRedox Titrationjeena josephОценок пока нет
- Homoeopathy and Minerals Jan Scholten.14414 1contents and ForewordДокумент6 страницHomoeopathy and Minerals Jan Scholten.14414 1contents and ForewordElanghovan Arumugam100% (1)
- Science8 - q3 - Mod4 - Periodic Table of Elements - v1Документ31 страницаScience8 - q3 - Mod4 - Periodic Table of Elements - v1Judarlyn MadriaОценок пока нет
- Surface Vehicle Recommended Practice: Rev. JUL95Документ10 страницSurface Vehicle Recommended Practice: Rev. JUL95raulОценок пока нет