Вам также может понравиться
- Production and Process ValidationДокумент60 страницProduction and Process ValidationdevsiddhidevОценок пока нет
- 2.03.04 Manu Sites Vaccine Prod ControlДокумент11 страниц2.03.04 Manu Sites Vaccine Prod ControlPrabhu LancerОценок пока нет
- Cleaning Validation PresentationДокумент66 страницCleaning Validation PresentationWali100% (1)
- Cleaning Validation: 1. PrincipleДокумент10 страницCleaning Validation: 1. PrincipleMadhusudan PanchalОценок пока нет
- Risk Based Environmental Monitoring ProgrammeДокумент51 страницаRisk Based Environmental Monitoring ProgrammeAugustine KoomsonОценок пока нет
- Microbiological Quality Control 2Документ37 страницMicrobiological Quality Control 2krbiotechОценок пока нет
- Session 2. Preparing For Cleaning ValidationДокумент49 страницSession 2. Preparing For Cleaning Validationrizka_yiyisОценок пока нет
- Cleaning Validation A Risk Based ApproacДокумент49 страницCleaning Validation A Risk Based ApproacOmnia GamalОценок пока нет
- 9 GMP OkДокумент104 страницы9 GMP Okleonny slОценок пока нет
- GMP & Process ValidationДокумент60 страницGMP & Process ValidationAhmed Zia100% (1)
- Cleaning Validation A Risk Based ApproachДокумент49 страницCleaning Validation A Risk Based Approachrodcam1100% (3)
- 2da Rev Anexo 1 PICs - Sterile - Medicinal - Products (20 Feb - 20may)Документ52 страницы2da Rev Anexo 1 PICs - Sterile - Medicinal - Products (20 Feb - 20may)Luis Gerardo Rendón BeltránОценок пока нет
- 3a CGMPДокумент73 страницы3a CGMPRICHELLE JIELEN QUEBEC100% (1)
- Current Good Manufacturing Practice & Drug Manufacturing QualityДокумент54 страницыCurrent Good Manufacturing Practice & Drug Manufacturing QualityGopinath GopiОценок пока нет
- Sanitary Design and Role of HACCP in SANITATION - HABUNGДокумент13 страницSanitary Design and Role of HACCP in SANITATION - HABUNGBhupen Hajong100% (1)
- QC Qa NotesДокумент66 страницQC Qa Noteskavya nainitaОценок пока нет
- Webinar Continuous Monitoring Concept - Final 1Документ18 страницWebinar Continuous Monitoring Concept - Final 1Trinh TrươngОценок пока нет
- 3 2 - MicrobiologicalQualityControl 1Документ37 страниц3 2 - MicrobiologicalQualityControl 1Tong ChanОценок пока нет
- PDAMicroEurope2018 TДокумент40 страницPDAMicroEurope2018 TNofrizalОценок пока нет
- APS For Cell TherapyДокумент19 страницAPS For Cell Therapydeepa_ragu5695Оценок пока нет
- Cleaning Validation Guidelines (GUIDE-0028) : Table of ContentsДокумент10 страницCleaning Validation Guidelines (GUIDE-0028) : Table of ContentsElitta MartinezОценок пока нет
- GMP TrainingДокумент14 страницGMP TrainingPrince MoniОценок пока нет
- Cleaning Validation in Pharmaceutical IndustryДокумент21 страницаCleaning Validation in Pharmaceutical IndustrysvengotoОценок пока нет
- CGMP Training Suntara Cosmetics Pvt. LTD.: Prepared By: Pratham ConsultantsДокумент76 страницCGMP Training Suntara Cosmetics Pvt. LTD.: Prepared By: Pratham Consultantsmrugeshj100% (1)
- 1 Sterile InterviewДокумент7 страниц1 Sterile Interviewabhishek.0dhaОценок пока нет
- KAP 3002 Chemical Safety Chater 1bДокумент37 страницKAP 3002 Chemical Safety Chater 1bSyahirah MisarihОценок пока нет
- PICs Annex1 Consultation Document Dec. 2017Документ50 страницPICs Annex1 Consultation Document Dec. 2017DholakiaОценок пока нет
- TRS 957 (2010) - Annex 3 - WHO GMP For Pharmaceutical Products Containing Hazardous SubstancesДокумент17 страницTRS 957 (2010) - Annex 3 - WHO GMP For Pharmaceutical Products Containing Hazardous SubstancesQuang Hiếu NgôОценок пока нет
- Anization, Personal and PremisesДокумент31 страницаAnization, Personal and PremisesNirvi ParekhОценок пока нет
- Bioburden Considerations in Equipment-Cleaning Validation: Did Not Include Swab Sampling of The Transfer LinesДокумент20 страницBioburden Considerations in Equipment-Cleaning Validation: Did Not Include Swab Sampling of The Transfer LinesWa Ode FasridaОценок пока нет
- Chapter 5 Production: Health and Consumers Directorate-General Public Health and Risk AssessmentДокумент11 страницChapter 5 Production: Health and Consumers Directorate-General Public Health and Risk AssessmentYasmeen ElyyanОценок пока нет
- Food Safety Best Practices For M/Smes (GMP & Haccp)Документ51 страницаFood Safety Best Practices For M/Smes (GMP & Haccp)Mark Nikko ManginsayОценок пока нет
- Presented By, Rasika Walunj M.Pharm (Qat) : Modern College of Pharmacy (For Ladies), Moshi PuneДокумент46 страницPresented By, Rasika Walunj M.Pharm (Qat) : Modern College of Pharmacy (For Ladies), Moshi PuneVivek PanchabhaiОценок пока нет
- CMC July 09 Patricia HughesДокумент30 страницCMC July 09 Patricia HughesSteven Correa MОценок пока нет
- Premises As Per GMPДокумент15 страницPremises As Per GMPdivvelaОценок пока нет
- Experiment No 3Документ3 страницыExperiment No 3laxmisalunke14Оценок пока нет
- Environmental Monitoring Considerations: Nancy Roscioli, Ph.D. Don Hill and Associates, IncДокумент54 страницыEnvironmental Monitoring Considerations: Nancy Roscioli, Ph.D. Don Hill and Associates, IncKandhakatla PraveenkumarОценок пока нет
- RESUMO Reprocessing-Medical-Devices-in-Health-Care-Settings - Validation-Methods-and-Labeling - Printable-SlidesДокумент42 страницыRESUMO Reprocessing-Medical-Devices-in-Health-Care-Settings - Validation-Methods-and-Labeling - Printable-Slidesluscafc caldasОценок пока нет
- Sanitary Design and Role of HACCP in SanitationДокумент12 страницSanitary Design and Role of HACCP in SanitationBhupen HajongОценок пока нет
- Food Processing EquipmentДокумент33 страницыFood Processing EquipmentplcuserОценок пока нет
- Guidelines For Routine Environmental Cleaning of The Operating RoomДокумент13 страницGuidelines For Routine Environmental Cleaning of The Operating RoomYnaffit Alteza UntalОценок пока нет
- CGMP Regulations of Sterile ProductsДокумент33 страницыCGMP Regulations of Sterile ProductsSukesh Potla75% (4)
- 4 Byron Burlingame 2Документ72 страницы4 Byron Burlingame 2watimelawatiОценок пока нет
- Prerequisite Programs (PRPS) Provide The Hygienic Foundations For Any Food Operation. The TermsДокумент6 страницPrerequisite Programs (PRPS) Provide The Hygienic Foundations For Any Food Operation. The TermsMawee LastrellaОценок пока нет
- Process ValidationДокумент16 страницProcess Validationahmed sharafОценок пока нет
- Food Safety - Focused Facility and Equipment Hygienic Design StrategiesДокумент6 страницFood Safety - Focused Facility and Equipment Hygienic Design StrategiesThẩm NguyễnОценок пока нет
- Env6 - Zoning Requirements, Pathogen Environment Monitoring For Dairy SuppliersДокумент37 страницEnv6 - Zoning Requirements, Pathogen Environment Monitoring For Dairy SuppliersMarianyОценок пока нет
- Microbiological Best Lab Practice&Environmental Monitoring-DikonversiДокумент26 страницMicrobiological Best Lab Practice&Environmental Monitoring-DikonversiNurul Hardiyanthi SadikinОценок пока нет
- HSA Cleaning Validation GuidelineДокумент11 страницHSA Cleaning Validation GuidelinesubirmeОценок пока нет
- Biorisk Management Basic-Level Materials: TD-O2-009: Biorisk Management Curriculum Development - Academic Track - JordanДокумент26 страницBiorisk Management Basic-Level Materials: TD-O2-009: Biorisk Management Curriculum Development - Academic Track - JordanMohammad Emad Al MadadhaОценок пока нет
- Kraft Allergen Management ForumДокумент63 страницыKraft Allergen Management ForumDavid100% (1)
- Good Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsОт EverandGood Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsОценок пока нет
- Effective microbiological sampling of food processing environments (1999)От EverandEffective microbiological sampling of food processing environments (1999)Оценок пока нет
- Biocontamination Control for Pharmaceuticals and HealthcareОт EverandBiocontamination Control for Pharmaceuticals and HealthcareРейтинг: 5 из 5 звезд5/5 (1)
- Cleaning and disinfection of food factories: a practical guideОт EverandCleaning and disinfection of food factories: a practical guideОценок пока нет
- Cleanroom Technology: Fundamentals of Design, Testing and OperationОт EverandCleanroom Technology: Fundamentals of Design, Testing and OperationОценок пока нет
- Carcass Management Guidelines: Effective Disposal of Animal Carcasses and Contaminated Materials on Small to Medium-Sized FarmsОт EverandCarcass Management Guidelines: Effective Disposal of Animal Carcasses and Contaminated Materials on Small to Medium-Sized FarmsОценок пока нет
- Quality Product ReviewДокумент10 страницQuality Product Reviewlina kharratОценок пока нет
- Temperature Mapping: Arcinova (CDMO)Документ6 страницTemperature Mapping: Arcinova (CDMO)lina kharratОценок пока нет
- Temperature Mapping: Arcinova (CDMO)Документ6 страницTemperature Mapping: Arcinova (CDMO)lina kharratОценок пока нет
- Report SanofiДокумент132 страницыReport Sanofilina kharratОценок пока нет
- The Regulatory Trends: Cross-Contamination in Drug ManufacturingДокумент9 страницThe Regulatory Trends: Cross-Contamination in Drug Manufacturinglina kharratОценок пока нет
- CLG418 (Dcec) PM 201409022-EnДокумент1 143 страницыCLG418 (Dcec) PM 201409022-EnMauricio WijayaОценок пока нет
- The Wayland News October 2014Документ16 страницThe Wayland News October 2014Julian HornОценок пока нет
- Civ Beyond Earth HotkeysДокумент1 страницаCiv Beyond Earth HotkeysExirtisОценок пока нет
- Acc116 Dec 2022 - Q - Test 1Документ6 страницAcc116 Dec 2022 - Q - Test 12022825274100% (1)
- Guardcam InstructionsДокумент12 страницGuardcam InstructionsCompuFix RepairsОценок пока нет
- 07 GDL Web-Site 04 (2021-2022) For 15284Документ2 страницы07 GDL Web-Site 04 (2021-2022) For 15284ABCDОценок пока нет
- ALE Manual For LaserScope Arc Lamp Power SupplyДокумент34 страницыALE Manual For LaserScope Arc Lamp Power SupplyKen DizzeruОценок пока нет
- Airport & Harbour Engg-AssignmentДокумент3 страницыAirport & Harbour Engg-AssignmentAshok Kumar RajanavarОценок пока нет
- Feed-Pump Hydraulic Performance and Design Improvement, Phase I: J2esearch Program DesignДокумент201 страницаFeed-Pump Hydraulic Performance and Design Improvement, Phase I: J2esearch Program DesignJonasОценок пока нет
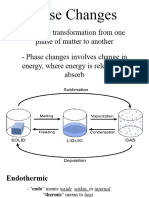
- General Chemistry 2 Q1 Lesson 5 Endothermic and Exotheric Reaction and Heating and Cooling CurveДокумент19 страницGeneral Chemistry 2 Q1 Lesson 5 Endothermic and Exotheric Reaction and Heating and Cooling CurveJolo Allexice R. PinedaОценок пока нет
- ADC of PIC MicrocontrollerДокумент4 страницыADC of PIC Microcontrollerkillbill100% (2)
- Participants ListДокумент13 страницParticipants Listmailway002Оценок пока нет
- Design and Analysis of Modified Front Double Wishbone Suspension For A Three Wheel Hybrid VehicleДокумент4 страницыDesign and Analysis of Modified Front Double Wishbone Suspension For A Three Wheel Hybrid VehicleRima AroraОценок пока нет
- User Manual PM3250Документ80 страницUser Manual PM3250otavioalcaldeОценок пока нет
- BMGT 200 Assignment 2 Answer KeysДокумент3 страницыBMGT 200 Assignment 2 Answer Keysharout keshishianОценок пока нет
- EKC 202ABC ManualДокумент16 страницEKC 202ABC ManualJose CencičОценок пока нет
- Top 100 Chemical CompaniesДокумент11 страницTop 100 Chemical Companiestawhide_islamicОценок пока нет
- FinalДокумент18 страницFinalAkash LadОценок пока нет
- Agency Canvas Ing PresentationДокумент27 страницAgency Canvas Ing Presentationkhushi jaiswalОценок пока нет
- Individual Career Plan: DIRECTIONS: Answer The Following Questions in Paragraph Form (3-4 Sentences) Per QuestionДокумент2 страницыIndividual Career Plan: DIRECTIONS: Answer The Following Questions in Paragraph Form (3-4 Sentences) Per Questionapi-526813290Оценок пока нет
- Lesson Plan For Implementing NETSДокумент5 страницLesson Plan For Implementing NETSLisa PizzutoОценок пока нет
- ETAP Power Station ErrorДокумент5 страницETAP Power Station ErroryogacruiseОценок пока нет
- I I I I: Peroxid.Q!Документ2 страницыI I I I: Peroxid.Q!Diego PradelОценок пока нет
- A Noble Noose of Methods - ExtendedДокумент388 страницA Noble Noose of Methods - ExtendedtomasiskoОценок пока нет
- School Based INSET Interim EvaluationДокумент8 страницSchool Based INSET Interim Evaluationprinces arcangelОценок пока нет
- P 348Документ196 страницP 348a123456978Оценок пока нет
- Segregation in CastingДокумент17 страницSegregation in CastingAsmaa Smsm Abdallh78% (9)
- Notes On Antibodies PropertiesДокумент3 страницыNotes On Antibodies PropertiesBidur Acharya100% (1)
- Introduction To BiogasДокумент5 страницIntroduction To BiogasLouis EldertardОценок пока нет
- Toh MFS8B 98B 003-11114-3AG1 PDFДокумент92 страницыToh MFS8B 98B 003-11114-3AG1 PDFDmitry NemtsoffОценок пока нет