Вам также может понравиться
- Introduccion A La Cocteleria PDFДокумент46 страницIntroduccion A La Cocteleria PDFAndrey 99Santy100% (2)
- Guía Del Instalador e Inspección para Conexiones Eléctricas CadweldДокумент30 страницGuía Del Instalador e Inspección para Conexiones Eléctricas CadweldRoberto Guzman SalinasОценок пока нет
- Taller 1 de Procesos IndustrialesДокумент2 страницыTaller 1 de Procesos IndustrialesSolcito DuránОценок пока нет
- Biosensores Aplicaciones en Los AlimentosДокумент11 страницBiosensores Aplicaciones en Los AlimentosJoseph ZamoraОценок пока нет
- Examen de Procesos IndustrialesДокумент3 страницыExamen de Procesos IndustrialesBenjamin Zambrana BazanОценок пока нет
- ENSAYO Aplicaciones Al Transporte de FluidoДокумент7 страницENSAYO Aplicaciones Al Transporte de FluidoCARMEN ROJASОценок пока нет
- Sulfuros, Halogenos Por Clasificación de StrunzДокумент11 страницSulfuros, Halogenos Por Clasificación de StrunzCristian Camilo Mejia HincapieОценок пока нет
- FICHA AMPLIACION T 2 Detective de Las SustanciasДокумент2 страницыFICHA AMPLIACION T 2 Detective de Las Sustanciasmelirod79Оценок пока нет
- Preparación de 100ml de Una Solución Estándar de Oxalato de Sodio Na2C2O4Документ4 страницыPreparación de 100ml de Una Solución Estándar de Oxalato de Sodio Na2C2O4Lorena Saenz FuentesОценок пока нет
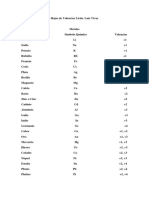
- Hojas de ValenciasДокумент6 страницHojas de ValenciasJesus SuarezОценок пока нет
- Precipitación PHДокумент5 страницPrecipitación PHantonioОценок пока нет
- Leyesdelosgases TPДокумент4 страницыLeyesdelosgases TPAgus RiosОценок пока нет
- Practica 1Документ12 страницPractica 1pamela pachecoОценок пока нет
- Clase9 Cap3 Clase6 Energía de GIBBS - Derivación y PropДокумент86 страницClase9 Cap3 Clase6 Energía de GIBBS - Derivación y PropDENNIS PABEL RAMIRO MAMANI SIMEONОценок пока нет
- Reacciones Analiticas de Nitrocompuestos PDFДокумент22 страницыReacciones Analiticas de Nitrocompuestos PDFANGELA MARIA VARGAS TABORDAОценок пока нет
- Taller 7 Enzimas - Estructura y FunciónДокумент6 страницTaller 7 Enzimas - Estructura y FunciónKatherin Juliana Guerrero NiñoОценок пока нет
- Taller 3 Semana 1Документ2 страницыTaller 3 Semana 1Brenda Quispe LescanoОценок пока нет
- Encuesta de Identificacion de PeligrosДокумент3 страницыEncuesta de Identificacion de PeligrosAURA DANIELA CASTRO AGUIRREОценок пока нет
- Quimica Organica III, EjerciciosДокумент2 страницыQuimica Organica III, EjerciciosValery.100% (1)
- Examen Parcial - Semana 4 - CB - SEGUNDO BLOQUE-FUNDAMENTOS DE QUIMICA - (GRUPO5) Res1 PDFДокумент5 страницExamen Parcial - Semana 4 - CB - SEGUNDO BLOQUE-FUNDAMENTOS DE QUIMICA - (GRUPO5) Res1 PDFPaola AZuñigaОценок пока нет
- Procesos de Union y Corte MetalesДокумент48 страницProcesos de Union y Corte MetalesJMiguel AqpОценок пока нет
- BibliografiaДокумент8 страницBibliografiaclaudiaОценок пока нет
- Planta MesapataДокумент19 страницPlanta MesapataAngel Mendoza100% (1)
- Problemario Tema 4 - Defectos Cristalinos - Parte 1Документ4 страницыProblemario Tema 4 - Defectos Cristalinos - Parte 1GiulianaОценок пока нет
- Estabilización de Suelo Unidad IIДокумент28 страницEstabilización de Suelo Unidad IIVictor Alfonso LaraОценок пока нет
- Tipo de TaladrosДокумент10 страницTipo de TaladrosEduardo TrejoОценок пока нет
- Termodinamica en Seres VivosДокумент2 страницыTermodinamica en Seres VivosEddy TrianaОценок пока нет
- Alcohol Isopropilico FDS QD 2018Документ10 страницAlcohol Isopropilico FDS QD 2018Mauricio Salgado LermandaОценок пока нет
- Guia de Aprendizaje Transporte de Sustancias Químicas PeligrosasДокумент14 страницGuia de Aprendizaje Transporte de Sustancias Químicas PeligrosasJaime Leonardo CruzОценок пока нет
- Nom 041 Semarnat 2015Документ35 страницNom 041 Semarnat 2015Uriel VazquezОценок пока нет