Вам также может понравиться
- Temperature-Responsive Polymers: Chemistry, Properties, and ApplicationsОт EverandTemperature-Responsive Polymers: Chemistry, Properties, and ApplicationsОценок пока нет
- Advanced Temperature Measurement and Control, Second EditionОт EverandAdvanced Temperature Measurement and Control, Second EditionОценок пока нет
- Polymerase Chain Reaction (PCR)Документ37 страницPolymerase Chain Reaction (PCR)Haleema SultanОценок пока нет
- PCR An OverviewДокумент41 страницаPCR An OverviewnassradabourОценок пока нет
- The Olymerase Hain Eaction: Zhang-Haifeng Department of BiochemistryДокумент42 страницыThe Olymerase Hain Eaction: Zhang-Haifeng Department of Biochemistryapi-19916399Оценок пока нет
- PCRДокумент11 страницPCRpkhkhawar100% (1)
- PCRДокумент42 страницыPCRNopiyana PujiastutiОценок пока нет
- Morgan 1922: 2000 Genes in 4 Chromosome of D.: MelanogasterДокумент44 страницыMorgan 1922: 2000 Genes in 4 Chromosome of D.: MelanogasterlordniklausОценок пока нет
- PCR ManualДокумент6 страницPCR ManualjiaОценок пока нет
- PCR Overview GoldBioДокумент1 страницаPCR Overview GoldBioKIARAH EUNIZE GICAОценок пока нет
- Polymerase Chain ReactionДокумент27 страницPolymerase Chain ReactionundalliОценок пока нет
- BB2111 PCR TOBEPRINTEDДокумент7 страницBB2111 PCR TOBEPRINTEDapi-3839553Оценок пока нет
- Polymerase Chain Reaction: Description of ComponentsДокумент5 страницPolymerase Chain Reaction: Description of ComponentsSamirSmsmОценок пока нет
- DNA Amplification 2020Документ5 страницDNA Amplification 2020Blameless ArikoОценок пока нет
- Polyme Rase Chain Reactio N: Course Code: 501Документ23 страницыPolyme Rase Chain Reactio N: Course Code: 501Humayun ArshadОценок пока нет
- The Polymerase Chain Reaction (PCR)Документ50 страницThe Polymerase Chain Reaction (PCR)Deana NamirembeОценок пока нет
- PCR LectureДокумент35 страницPCR LectureArfan Tri KusumaОценок пока нет
- PCR Protocol For Taq DNA Polymerase With Standard Taq BufferДокумент2 страницыPCR Protocol For Taq DNA Polymerase With Standard Taq BufferAnonymous DA8iQzОценок пока нет
- Polymerase Chain ReactionДокумент14 страницPolymerase Chain ReactionDavidMugambi100% (1)
- ArticleДокумент3 страницыArticleSanggari AnamalaiОценок пока нет
- Polymerase Chain Reaction: Salwa Hassan TeamaДокумент56 страницPolymerase Chain Reaction: Salwa Hassan TeamaSamiksha SharmaОценок пока нет
- 03 PCRДокумент5 страниц03 PCRToni D.Оценок пока нет
- Igem Training 1Документ45 страницIgem Training 1api-343562611Оценок пока нет
- Module 4 - PCRДокумент12 страницModule 4 - PCRnavneetОценок пока нет
- PCRДокумент34 страницыPCRSumitОценок пока нет
- Polymerase Chain Reaction ObjectivesДокумент25 страницPolymerase Chain Reaction ObjectivestinasheОценок пока нет
- Polymerase Chain Reaction: Description of ComponentsДокумент5 страницPolymerase Chain Reaction: Description of ComponentsSkand BhardwajОценок пока нет
- PCR Polymerase Chain Reaction PPT 1 1Документ106 страницPCR Polymerase Chain Reaction PPT 1 1MaithiliОценок пока нет
- Prerequisite of A PCR: Logarithmic and Controlled FashionДокумент18 страницPrerequisite of A PCR: Logarithmic and Controlled FashionlkokodkodОценок пока нет
- Primer Designing For PCRДокумент13 страницPrimer Designing For PCRiftikharsubha5Оценок пока нет
- KOD Hot StartДокумент4 страницыKOD Hot Startsike1977Оценок пока нет
- Polymerase Chain ReactionДокумент47 страницPolymerase Chain ReactionshvnagaОценок пока нет
- TYPES OF PCR - Dr. De-1 PDFДокумент33 страницыTYPES OF PCR - Dr. De-1 PDFJAICHANDRUОценок пока нет
- Polymerase Chain Reaction (PCR)Документ21 страницаPolymerase Chain Reaction (PCR)greateinsteinОценок пока нет
- PCR RTPCR, QPCRДокумент88 страницPCR RTPCR, QPCRshravani sahuОценок пока нет
- Condiciones PCR Con AmplitaqДокумент6 страницCondiciones PCR Con AmplitaqSakura JunОценок пока нет
- Different Types of Polymerase Chain ReactionДокумент7 страницDifferent Types of Polymerase Chain ReactionKristine Ann100% (1)
- The Polymerase Chain Reaction (PCR) : BackgroundДокумент4 страницыThe Polymerase Chain Reaction (PCR) : BackgroundМ.К. МариОценок пока нет
- Polymerase Chain ReactionДокумент39 страницPolymerase Chain ReactionNurul FatikhaОценок пока нет
- PCR Protocol For Taq DNA Polymerase With Standard Taq Buffer (M0273)Документ3 страницыPCR Protocol For Taq DNA Polymerase With Standard Taq Buffer (M0273)Despoina ChatziОценок пока нет
- Polymerase Chain ReactionДокумент60 страницPolymerase Chain Reactionclarice_anneОценок пока нет
- Bio Project-Polymerase Chain ReactionДокумент21 страницаBio Project-Polymerase Chain ReactionS.AbiniveshОценок пока нет
- Analiza PCR - ThermocyclerДокумент7 страницAnaliza PCR - Thermocyclerionescu_ion_3Оценок пока нет
- Maria Afzal M.phill-03 Submitted To: Prof. Dr. Farkhanda JabeenДокумент44 страницыMaria Afzal M.phill-03 Submitted To: Prof. Dr. Farkhanda JabeenMuhammad MushtaqОценок пока нет
- Molecular Diagnostics Techniques Polymerase Chain ReactionДокумент7 страницMolecular Diagnostics Techniques Polymerase Chain ReactionJamesОценок пока нет
- Introduction PCR AssignmentДокумент20 страницIntroduction PCR AssignmentogunfunminiyiestherОценок пока нет
- Ex No.-03 Polymerase Chain ReactionДокумент4 страницыEx No.-03 Polymerase Chain ReactionSujit ShandilyaОценок пока нет
- Principle of The PCR: This Technique Is Invented in 1984 by Kary Mulis. The Purpose of A PCRДокумент11 страницPrinciple of The PCR: This Technique Is Invented in 1984 by Kary Mulis. The Purpose of A PCRantesar reheemОценок пока нет
- Lecture 3 Diagnostic GenomicsДокумент40 страницLecture 3 Diagnostic Genomicssales zfОценок пока нет
- KOD Hot Start PolymeraseДокумент8 страницKOD Hot Start PolymeraseJen LeiОценок пока нет
- Protocolo One Taq PolimerasaДокумент5 страницProtocolo One Taq PolimerasaJenifer Castro EstradaОценок пока нет
- PCR Primer DesignДокумент13 страницPCR Primer DesignAiman ArshadОценок пока нет
- Dream Taq Green MM #k1081Документ2 страницыDream Taq Green MM #k1081victor_david_19Оценок пока нет
- JumpStart REDTaq Ready MixДокумент4 страницыJumpStart REDTaq Ready MixHarrison WangОценок пока нет
- Polymerase Chain ReactionДокумент34 страницыPolymerase Chain Reactionmokshgoyal2597100% (4)
- Polymerase Chain ReactionДокумент13 страницPolymerase Chain Reactionahmad ghОценок пока нет
- DR Noha PCRДокумент62 страницыDR Noha PCRsaged loaiОценок пока нет
- DNA Amplification and PCR: by Sara Haydar Mohammed Hasan Fourth Stage A2 Morning StudyДокумент9 страницDNA Amplification and PCR: by Sara Haydar Mohammed Hasan Fourth Stage A2 Morning Studydaloo dmdmОценок пока нет
- Polymerase Chain ReactionДокумент39 страницPolymerase Chain ReactionShefali PawarОценок пока нет
- Go-Taq Long PCR Master Mix ProtocolДокумент10 страницGo-Taq Long PCR Master Mix ProtocolZОценок пока нет
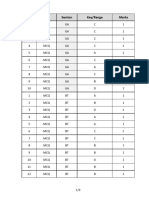
- Q.No. Type Section Key/Range MarksДокумент3 страницыQ.No. Type Section Key/Range Marksbiotech_vidhyaОценок пока нет
- Nuclear ExtractsДокумент2 страницыNuclear Extractsbiotech_vidhyaОценок пока нет
- BT 2019Документ13 страницBT 2019biotech_vidhyaОценок пока нет
- Troubleshooting SDS-PAGE 1Документ3 страницыTroubleshooting SDS-PAGE 1biotech_vidhyaОценок пока нет
- Buffer Preparation PDFДокумент6 страницBuffer Preparation PDFbiotech_vidhyaОценок пока нет
- Ies 17 Set A Me Q AДокумент67 страницIes 17 Set A Me Q Abiotech_vidhyaОценок пока нет
- TNPSC Group 1 Prelim Book List PDFДокумент2 страницыTNPSC Group 1 Prelim Book List PDFbiotech_vidhyaОценок пока нет
- Whole Cell ExtractДокумент1 страницаWhole Cell Extractbiotech_vidhyaОценок пока нет
- Mechanical Engineering Code No. 14: Combined Competitive (Preliminary) Examination, 2010Документ20 страницMechanical Engineering Code No. 14: Combined Competitive (Preliminary) Examination, 2010biotech_vidhyaОценок пока нет
- Befcv List PDFДокумент22 страницыBefcv List PDFbiotech_vidhyaОценок пока нет
- Isa BusДокумент30 страницIsa Busbiotech_vidhya100% (1)
- Lec09 Three PhaseДокумент84 страницыLec09 Three Phasebiotech_vidhyaОценок пока нет
- 1 TolerancesДокумент1 страница1 Tolerancesbiotech_vidhyaОценок пока нет
- Teachers Recruitment Board: 1. Important DatesДокумент13 страницTeachers Recruitment Board: 1. Important Datesbiotech_vidhyaОценок пока нет
- PCR and Agarose Gel ElectrophoresisДокумент5 страницPCR and Agarose Gel ElectrophoresisEamon Barkhordarian100% (1)
- Edwin Darmawan: Moderator: Dr. Dr. Hani Susianti, SP - PK (K)Документ48 страницEdwin Darmawan: Moderator: Dr. Dr. Hani Susianti, SP - PK (K)Edwin DarmawanОценок пока нет
- Genei: Student PCR Teaching Kit ManualДокумент13 страницGenei: Student PCR Teaching Kit ManualSoma GhoshОценок пока нет
- DNA Sequencing: Kabi R. Neupane, Ph.D. Leeward Community College ABE Workshop 2006Документ17 страницDNA Sequencing: Kabi R. Neupane, Ph.D. Leeward Community College ABE Workshop 2006ximenalrОценок пока нет
- Guide To Electropherogram v3Документ12 страницGuide To Electropherogram v3varveroonОценок пока нет
- Chapter 2 Literature ReviewДокумент30 страницChapter 2 Literature ReviewSemana HumedalesОценок пока нет
- Genetics Lab ManualДокумент90 страницGenetics Lab ManualRawan Al AbbassiОценок пока нет
- Long Essay 2Документ7 страницLong Essay 2Alexandru-Crivac Cristina-NicoletaОценок пока нет
- KOD Xtreme™ Hot Start DNA PolymeraseДокумент8 страницKOD Xtreme™ Hot Start DNA Polymerase1pisco1Оценок пока нет
- Recombinant Dna Technology & It's ApplicationДокумент20 страницRecombinant Dna Technology & It's Applicationiamcold866Оценок пока нет
- PCR Alupv92 09Документ19 страницPCR Alupv92 09asiah sallehОценок пока нет
- G9 STE Q3 Biotechnology ReviewerДокумент9 страницG9 STE Q3 Biotechnology ReviewerJoyz Etneilas ArguillesОценок пока нет
- Forensic Trace DNA: A ReviewДокумент17 страницForensic Trace DNA: A ReviewAyu AndrianОценок пока нет
- Q5 Site-Directed Mutagenesis Kit (Without Competent Cells) : Dna Modifying EnzymesДокумент16 страницQ5 Site-Directed Mutagenesis Kit (Without Competent Cells) : Dna Modifying EnzymesSusan HsiaoОценок пока нет
- Molecular Biology Notes 1 PDFДокумент23 страницыMolecular Biology Notes 1 PDFChris_Barber09100% (3)
- ADP 23 Long-Range PCR Rev BДокумент16 страницADP 23 Long-Range PCR Rev BClaire FieldingОценок пока нет
- Departments: SDMVM'S College of Agricultural BiotechnologyДокумент45 страницDepartments: SDMVM'S College of Agricultural BiotechnologyPAWANKUMAR S. K.Оценок пока нет
- Isolation and Characterization of Rhizobacteria From ChilliДокумент15 страницIsolation and Characterization of Rhizobacteria From ChillisauravОценок пока нет
- Castes and Tribes in Hazara Division, Khyber Pakhtunkhwa, PakistanДокумент11 страницCastes and Tribes in Hazara Division, Khyber Pakhtunkhwa, PakistanScribd UploadsОценок пока нет
- Mytaq Hs Red Mix Product ManualДокумент2 страницыMytaq Hs Red Mix Product ManualDita Dwi Rahma100% (1)
- Lab 09 - Mendelian Genetics & PCRДокумент20 страницLab 09 - Mendelian Genetics & PCRBryanОценок пока нет
- DNA Sequencing (Article) - Biotechnology - Khan AcademyДокумент8 страницDNA Sequencing (Article) - Biotechnology - Khan AcademyZannat AraОценок пока нет
- PCR Questions and Answers Sep PagesДокумент5 страницPCR Questions and Answers Sep PagesMohita Chugh100% (1)
- Sensors and Actuators B: Chemical: Syahidah Nurani Zulki I, Herlina Abdul Rahim, Woei-Jye LauДокумент33 страницыSensors and Actuators B: Chemical: Syahidah Nurani Zulki I, Herlina Abdul Rahim, Woei-Jye LauAlejandra QuintinОценок пока нет
- Setting Up A PCR LaboratoryДокумент11 страницSetting Up A PCR LaboratoryMet RizalОценок пока нет
- Genetics CancerДокумент206 страницGenetics CancerDr. Serin KuriakoseОценок пока нет
- Primer Design ExerciseДокумент34 страницыPrimer Design ExerciseRita karen Pacheco CabañasОценок пока нет
- EN QuantiTect Reverse Transcription Handbook PDFДокумент32 страницыEN QuantiTect Reverse Transcription Handbook PDFFelp ScholzОценок пока нет
- Bio MEMSДокумент52 страницыBio MEMSShobhit SinghОценок пока нет
- Ion Torrent Technology and WorkflowДокумент33 страницыIon Torrent Technology and Workflowpham trang100% (1)