Вам также может понравиться
- Processus irréversibles non linéaires en thermodynamique: Les Grands Articles d'UniversalisОт EverandProcessus irréversibles non linéaires en thermodynamique: Les Grands Articles d'UniversalisОценок пока нет
- Exercices TABLEAUX A TERMINER PDFДокумент28 страницExercices TABLEAUX A TERMINER PDFAnou ArОценок пока нет
- GénéralitésДокумент5 страницGénéralitéspamy26250Оценок пока нет
- GNL 2 PDFДокумент133 страницыGNL 2 PDFعبد الحميد الجزائريОценок пока нет
- CL À Simple Battant Awastop B680 PDFДокумент4 страницыCL À Simple Battant Awastop B680 PDFsoufitoutiОценок пока нет
- Guide Utilisation Dealer MessenkaДокумент25 страницGuide Utilisation Dealer MessenkaYANN MAMBOОценок пока нет
- Fiches Clap PDFДокумент253 страницыFiches Clap PDFsebastienschlesserОценок пока нет
- Cours SP2013Документ65 страницCours SP2013lafaffОценок пока нет
- Controle ComandeДокумент73 страницыControle ComandeRnav rnavОценок пока нет
- Les Fluides Utilises en Genie FrigorifiqueДокумент12 страницLes Fluides Utilises en Genie FrigorifiquepeziereОценок пока нет
- Tpe Carateristique Des ReservoirsДокумент11 страницTpe Carateristique Des Reservoirstanda notsawoОценок пока нет
- Specification Technique Pompe Et Plan Qualite PDFДокумент16 страницSpecification Technique Pompe Et Plan Qualite PDFخربوش سماعينОценок пока нет
- l1.1 Varedia Livret TechnologiesДокумент46 страницl1.1 Varedia Livret TechnologiesJàMàl MejorОценок пока нет
- THERMODYДокумент61 страницаTHERMODYKhalid DeliegeОценок пока нет
- Bâches Dégazantes Babcock-WansonДокумент2 страницыBâches Dégazantes Babcock-WansonbaptОценок пока нет
- Dai Fichesureindustrie Reacteurschimiques PDFДокумент24 страницыDai Fichesureindustrie Reacteurschimiques PDFمحمد الأمير الحازميОценок пока нет
- Piping PlanДокумент54 страницыPiping PlanichrakОценок пока нет
- Regulflex Refrigeration Aliment A Ire ERM FRДокумент10 страницRegulflex Refrigeration Aliment A Ire ERM FRMohamed Amine JazouliОценок пока нет
- Cours Ingénierie Des FLNG 2022-2023 - Chap2-V1Документ86 страницCours Ingénierie Des FLNG 2022-2023 - Chap2-V1Franklin FossokОценок пока нет
- Petrochimie 2Документ57 страницPetrochimie 2Oüssàma Khęllįl100% (1)
- 1213 Thermodynamique PhysiqueДокумент5 страниц1213 Thermodynamique Physiqueridhajamel0% (1)
- Niveau XДокумент18 страницNiveau XTouil AmirОценок пока нет
- Gaz TorchésДокумент18 страницGaz Torchéswissoubenaouda2110Оценок пока нет
- Procedure Demarrage MCRДокумент18 страницProcedure Demarrage MCRSofiane HalimiОценок пока нет
- Boil Off Gas PDFДокумент1 страницаBoil Off Gas PDFNina KrnhОценок пока нет
- 1 Actionneurs de R Gulation Vanne PDFДокумент19 страниц1 Actionneurs de R Gulation Vanne PDFDODOHICHAMОценок пока нет
- Hibon - BlowerДокумент8 страницHibon - BlowerSergio IvánОценок пока нет
- Chapitre IДокумент12 страницChapitre ILkl HadjerОценок пока нет
- B002 Directive Qualité de Leau LOOS MaJ 112014Документ16 страницB002 Directive Qualité de Leau LOOS MaJ 112014Jero MilОценок пока нет
- T.P. FLUENT. Cours Mécanique Des Fluides. 24 Février 2006 NAZIH MARZOUQYДокумент31 страницаT.P. FLUENT. Cours Mécanique Des Fluides. 24 Février 2006 NAZIH MARZOUQYbebelcire0% (1)
- PFE BensaciДокумент17 страницPFE Bensacisyd d-waterlawОценок пока нет
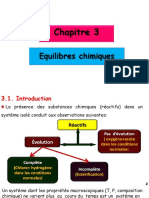
- Chapitre 3 Equilibres Chimiques 3ème Génie Des ProcédésДокумент23 страницыChapitre 3 Equilibres Chimiques 3ème Génie Des ProcédésCherif LaifaОценок пока нет
- 9 - Machine Tournantes Rev 2005Документ156 страниц9 - Machine Tournantes Rev 2005mountsoОценок пока нет
- Catalog Recondi Sectoriel 2015Документ280 страницCatalog Recondi Sectoriel 2015atisz333Оценок пока нет
- Distributeur EleveДокумент2 страницыDistributeur EleveHafedh HammamiОценок пока нет
- Le Guide de La Chaudière À Condensation Gaz PDFДокумент3 страницыLe Guide de La Chaudière À Condensation Gaz PDFLahouari FatahОценок пока нет
- Chetoui - Zineddine Ferjdani - Racim-RifaatДокумент93 страницыChetoui - Zineddine Ferjdani - Racim-RifaatHadj MeknassiОценок пока нет
- Exercice AzoteДокумент4 страницыExercice Azoteabahri AThmaneОценок пока нет
- Application Notes Interfacing DEIF Equipment 4189340670 FRДокумент47 страницApplication Notes Interfacing DEIF Equipment 4189340670 FRAziz ArrahalОценок пока нет
- Simulateur de Procidé Chapitre1Документ29 страницSimulateur de Procidé Chapitre1HadjОценок пока нет
- Bruleur Chaudiere A EauДокумент36 страницBruleur Chaudiere A Eausami ben chikhaОценок пока нет
- Rapport de Stage SONELДокумент62 страницыRapport de Stage SONELGuy EffaОценок пока нет
- Le Rapport Ec141 NouveuxДокумент23 страницыLe Rapport Ec141 Nouveuxعبد القادر دربالОценок пока нет
- Comptage Thermique Vapeur PDFДокумент26 страницComptage Thermique Vapeur PDFOmar RajhiОценок пока нет
- C7-2 BДокумент15 страницC7-2 BamineОценок пока нет
- Institut Algérien Du PétroleДокумент55 страницInstitut Algérien Du PétroleSofiane Halimi100% (2)
- SystemeschauffageДокумент30 страницSystemeschauffageyaxinoОценок пока нет
- Webinar H2 29092020 PresentationДокумент31 страницаWebinar H2 29092020 PresentationShare Point100% (1)
- L'impact de La Filtration de L'air D'admission Sur Le Rendement de La Turbine À Gaz MS5002CДокумент78 страницL'impact de La Filtration de L'air D'admission Sur Le Rendement de La Turbine À Gaz MS5002Cabdelhakim100% (1)
- Adduction en Eau Et CanalisationДокумент52 страницыAdduction en Eau Et CanalisationYvanОценок пока нет
- Grace 2Документ13 страницGrace 2Jonas lumbuОценок пока нет
- Sonatrach Raffinerie D'algerДокумент89 страницSonatrach Raffinerie D'algerSoundous TayssirОценок пока нет
- Guide Lotisseurs Eau 2012Документ104 страницыGuide Lotisseurs Eau 2012ihsan hassouneОценок пока нет
- Exemples de SimulationДокумент172 страницыExemples de SimulationoussamaОценок пока нет
- IntroductionДокумент5 страницIntroductionIlyes BenhaceneОценок пока нет
- Réseau VapeurДокумент26 страницRéseau VapeurPaul Julbin NJOCKОценок пока нет
- TD 3Документ11 страницTD 3Abderrahim SaifiОценок пока нет
- Thermodynamique Chimique: Cours Du ModuleДокумент115 страницThermodynamique Chimique: Cours Du ModuleG95 PfeОценок пока нет
- Master Thermo 2Документ33 страницыMaster Thermo 2Mohamed EL FAGHLOUMIОценок пока нет
- (CHAPITRE I RECTF) Généralité Sur La Thermodynamique PDFДокумент22 страницы(CHAPITRE I RECTF) Généralité Sur La Thermodynamique PDFRamzi OunisОценок пока нет
- Cours de Geologie PetroliereДокумент43 страницыCours de Geologie Petroliereazul93% (15)
- 36 Hydrocarbures Génie GazierДокумент43 страницы36 Hydrocarbures Génie GazierKHALEDFEKAIRОценок пока нет
- Proprietes PVTДокумент18 страницProprietes PVTKHALEDFEKAIR100% (1)
- EnergyДокумент6 страницEnergyKHALEDFEKAIRОценок пока нет
- KhodjaДокумент288 страницKhodjaAbdelmoneme Jaballah50% (2)
- Hadjadj Hadjoudj PDFДокумент63 страницыHadjadj Hadjoudj PDFKHALEDFEKAIRОценок пока нет
- Kha5791 PDFДокумент165 страницKha5791 PDFKHALEDFEKAIRОценок пока нет
- GDF SonatrachДокумент39 страницGDF SonatrachKHALEDFEKAIRОценок пока нет
- 02 GazReels PDFДокумент37 страниц02 GazReels PDFKHALEDFEKAIRОценок пока нет
- Felioune Kouidri PDFДокумент73 страницыFelioune Kouidri PDFKHALEDFEKAIR100% (1)
- Liquefaction D'un Gaz PDFДокумент15 страницLiquefaction D'un Gaz PDFKHALEDFEKAIRОценок пока нет
- Emociones (Roberto Carlos) Eb - VioloncelloДокумент1 страницаEmociones (Roberto Carlos) Eb - VioloncelloRoberto Aguilar EscobedoОценок пока нет
- CODE DE LA SECURITE INTERIEURE Correspondance Loi 83 629 PDFДокумент24 страницыCODE DE LA SECURITE INTERIEURE Correspondance Loi 83 629 PDFmig972972Оценок пока нет
- Introitus Apertus Ad Occlusum Regis (... ) Philalethes Eugenius bpt6k746977Документ449 страницIntroitus Apertus Ad Occlusum Regis (... ) Philalethes Eugenius bpt6k746977MannekkeОценок пока нет
- CompteДокумент11 страницCompteJehdhd VdhdhdhОценок пока нет
- Recepisse CR 4Документ1 страницаRecepisse CR 4api-269179615Оценок пока нет
- LA CUICHI - BANDA MM Version 2Документ15 страницLA CUICHI - BANDA MM Version 2German Manzano100% (1)
- Rapport Métier Et FormationДокумент18 страницRapport Métier Et FormationAchraf AZ Zouitina78% (18)
- DROIT DES OBLIGATIONS - CAS PRATIQUE ExercieДокумент5 страницDROIT DES OBLIGATIONS - CAS PRATIQUE Exerciecissokho1900Оценок пока нет
- Der Ursprung Der Moralischen Empfindungen (... ) Rée Paul bpt6k943536 PDFДокумент158 страницDer Ursprung Der Moralischen Empfindungen (... ) Rée Paul bpt6k943536 PDFLeonel AntunesОценок пока нет
- M03 Gestion urbaine-BTP-TRBT PDFДокумент42 страницыM03 Gestion urbaine-BTP-TRBT PDFMohamed KanzoutОценок пока нет
- Contrat de Commission Sur Vente International AnglaisДокумент6 страницContrat de Commission Sur Vente International AnglaisAliou CisseОценок пока нет
- Code Maçonnique DesДокумент22 страницыCode Maçonnique DesRoberto Amato100% (1)
- Migne. Patrologia Latina Tomus CVIII.Документ619 страницMigne. Patrologia Latina Tomus CVIII.Patrologia Latina, Graeca et OrientalisОценок пока нет
- Sujet 1 Et Corrigé-1Документ4 страницыSujet 1 Et Corrigé-1cyrilОценок пока нет
- FRAGIL - Trompeta en SibДокумент2 страницыFRAGIL - Trompeta en Sibektor martinОценок пока нет
- Conditions Générales de Prestation de Services OkДокумент4 страницыConditions Générales de Prestation de Services OkBalakankatharan PrashanaОценок пока нет
- Ka 3 BechДокумент65 страницKa 3 BechClay IipОценок пока нет
- Sevillanas (Manuel Pareja Obregón Arr Carlos Guillén) - PianoДокумент3 страницыSevillanas (Manuel Pareja Obregón Arr Carlos Guillén) - PianoEmilio Damián Guerrero Sánchez100% (1)
- THIOYE - Droit Des Affaires - Corps 3Документ42 страницыTHIOYE - Droit Des Affaires - Corps 3LucasОценок пока нет
- No Hubo Un Heroe Más Grande - Kenneth CopeДокумент8 страницNo Hubo Un Heroe Más Grande - Kenneth CopePablo CastilloОценок пока нет
- Hs 4Документ78 страницHs 4bookmolen_60709275Оценок пока нет
- Credit BailДокумент13 страницCredit BailLounes AlilatОценок пока нет
- Accord de Confidencialité - ModèleДокумент1 страницаAccord de Confidencialité - ModèleOrlindo WagnerОценок пока нет
- Biology Paper 1 SL SpanishДокумент17 страницBiology Paper 1 SL SpanishPrisila CastroОценок пока нет
- Imm0008 4f PDFДокумент1 страницаImm0008 4f PDFberteОценок пока нет
- Pass CamДокумент1 страницаPass CamGazali MohamedОценок пока нет
- CoursДокумент19 страницCoursAbergelОценок пока нет
- Question Du Juge Naturel en Droit Congolais Justice Congolaise2Документ13 страницQuestion Du Juge Naturel en Droit Congolais Justice Congolaise2Exauce Zess NdombaОценок пока нет
- Auto-Certification de Résidence Fiscale Personne MoraleДокумент4 страницыAuto-Certification de Résidence Fiscale Personne MoralePHILIPPE DIABANОценок пока нет
- Journal El Watan Du Vendredi 9 Fevrier 2018Документ24 страницыJournal El Watan Du Vendredi 9 Fevrier 2018Stukine100% (1)