Вам также может понравиться
- Evaluación e Intervención Neuropsicológica en Un Caso de Demencia Por Enfermedad de Parkinson PDFДокумент42 страницыEvaluación e Intervención Neuropsicológica en Un Caso de Demencia Por Enfermedad de Parkinson PDFMARIANA LEIVA ZÚÑIGAОценок пока нет
- Sindrome de Prader WillyДокумент10 страницSindrome de Prader WillymatiasОценок пока нет
- MIASTENIA GRAVIS Investigacion NeuroДокумент12 страницMIASTENIA GRAVIS Investigacion NeuroOskhar Castillo ZambranoОценок пока нет
- GRUPO 2 Otorrino 10 Casos 1.0Документ32 страницыGRUPO 2 Otorrino 10 Casos 1.0NALINE GUSMAO LOPES LIMAОценок пока нет
- Monocorditis VasomotoraДокумент8 страницMonocorditis Vasomotoralisandri sanabriaОценок пока нет
- DegluciónДокумент51 страницаDegluciónNatalie DanielaОценок пока нет
- Caso ClínicoДокумент28 страницCaso ClínicoAmarilis Palacios100% (1)
- Evaluacion de Deglución E.G.Документ2 страницыEvaluacion de Deglución E.G.Carolina Andrea Vasquez HenriquezОценок пока нет
- Síndrome VelocardiofacialДокумент24 страницыSíndrome VelocardiofacialJennifer MettigОценок пока нет
- AlimentacionComplementariaGuiadaPorElBebe 4847927Документ9 страницAlimentacionComplementariaGuiadaPorElBebe 4847927lendhorОценок пока нет
- Disfagia Paralisis CerebralДокумент33 страницыDisfagia Paralisis CerebralMoniUcmОценок пока нет
- TERAPIAsДокумент15 страницTERAPIAsNayareth Marambio OrellanaОценок пока нет
- Tratamiento de Los Trastornos de La DegluciónДокумент43 страницыTratamiento de Los Trastornos de La DegluciónAlgutcaОценок пока нет
- Deglución - CamporaДокумент7 страницDeglución - CamporaKAREN LIGUORIОценок пока нет
- Intervencion Fonoaudiologica en Pctes Con Fisura LabiopalatinaДокумент14 страницIntervencion Fonoaudiologica en Pctes Con Fisura LabiopalatinaJuanjoCorderoОценок пока нет
- Patologías CДокумент3 страницыPatologías Cmaria vОценок пока нет
- Síndrome de Prader - WillyДокумент7 страницSíndrome de Prader - WillyMaria BahamondeОценок пока нет
- DemenciaДокумент3 страницыDemenciaEstefania Milena PimentelОценок пока нет
- Bases Neurobiológicas de Deglución Trastornos y TratamientoДокумент22 страницыBases Neurobiológicas de Deglución Trastornos y TratamientoCamila CarreñoОценок пока нет
- Lesiones Hemisferio DerechoДокумент4 страницыLesiones Hemisferio DerechoLesli Moreno Pérez100% (1)
- Cuidado y Rehabilitación Paciente TraqueotomizadoДокумент79 страницCuidado y Rehabilitación Paciente TraqueotomizadoSandra Garriga GalobardesОценок пока нет
- EnvejecimientoДокумент25 страницEnvejecimientoplt090Оценок пока нет
- Efectos de Miastenia Gravis en La Voz, El Habla y Al TragarДокумент2 страницыEfectos de Miastenia Gravis en La Voz, El Habla y Al TragarPily Barde AvilaОценок пока нет
- DisartriaДокумент11 страницDisartriaSonia Palma100% (1)
- Caso Clínico Del Trastorno Del LenguajeДокумент9 страницCaso Clínico Del Trastorno Del LenguajeJILARY BRITNEY CASTILLO RUIZОценок пока нет
- TCC DemenciasДокумент26 страницTCC DemenciasFlga Claudia Ponce FloresОценок пока нет
- Caso Clínico SPW - AlissaДокумент19 страницCaso Clínico SPW - AlissaAlissa Ivonne Rodriguez LeonesОценок пока нет
- Afasia Motora Eferente o de BrocaДокумент15 страницAfasia Motora Eferente o de BrocaKaty LuuОценок пока нет
- Dr. Rafael Cotos Canca PDFДокумент99 страницDr. Rafael Cotos Canca PDFAngelica GarciaОценок пока нет
- Protocolo Exploración Inicial Niños y AdolescentesДокумент2 страницыProtocolo Exploración Inicial Niños y Adolescentesdanitza san martin100% (1)
- Caso Clinico Miopatia NemalinicaДокумент6 страницCaso Clinico Miopatia NemalinicaAndi Monasterios TejerinaОценок пока нет
- Disfagia OrofaringeaДокумент16 страницDisfagia OrofaringeaJohanita E. Mendez100% (1)
- DISARTRIASДокумент3 страницыDISARTRIASDanielaSayagoRuizОценок пока нет
- Disfagia en La Esclerosis MúltipleДокумент15 страницDisfagia en La Esclerosis MúltipleMilenOjedaОценок пока нет
- Profe Pablo, Maniobras de LiberacionДокумент6 страницProfe Pablo, Maniobras de LiberacionValentina TreceОценок пока нет
- Fisiología Aplicada A Vía AéreaДокумент5 страницFisiología Aplicada A Vía AéreabarbaraОценок пока нет
- La DisfagiaДокумент15 страницLa DisfagiaVirginia GimenezОценок пока нет
- PrebifagiaДокумент5 страницPrebifagiaValentina Paz Fuentes Cáceres100% (1)
- Enfermedad de Meniere PDFДокумент7 страницEnfermedad de Meniere PDFAriana VadoОценок пока нет
- Fisura Labio-Palatina (Dr. Mendiguri) Grupo 4Документ40 страницFisura Labio-Palatina (Dr. Mendiguri) Grupo 4Jarol Justo MamaniОценок пока нет
- OrtesisДокумент4 страницыOrtesispaulinacorpalОценок пока нет
- Deglucion en El Adulto MayorДокумент7 страницDeglucion en El Adulto MayorKaty VwОценок пока нет
- Cuadro Comparativo Demencia Nutricional y ParkinsonДокумент2 страницыCuadro Comparativo Demencia Nutricional y ParkinsonMahatma IzaОценок пока нет
- Disfagia Orofaríngea Como Un Nuevo Síndrome GeriátricoДокумент4 страницыDisfagia Orofaríngea Como Un Nuevo Síndrome GeriátricoJavier WafflesОценок пока нет
- Fundamentos De: ArtrogriposisДокумент12 страницFundamentos De: ArtrogriposisRisa AleОценок пока нет
- Ataxia de Friedrich 1Документ3 страницыAtaxia de Friedrich 1SOAD123Оценок пока нет
- Graficación VPPBДокумент11 страницGraficación VPPBNatalia Stefanny MuñozОценок пока нет
- VPPBДокумент31 страницаVPPBValeria Bahamondez RuizОценок пока нет
- Sindromes NeurológicosДокумент66 страницSindromes NeurológicosAbigail GallardoОценок пока нет
- Caso Clinico 4Документ1 страницаCaso Clinico 4Fran GuerreroОценок пока нет
- Rehabilitación en Pacientes Con TECДокумент12 страницRehabilitación en Pacientes Con TECJavier Alejandro Saravia SeoaneОценок пока нет
- Otitis Media Con Efusión y Fibroadhesiva 2019Документ10 страницOtitis Media Con Efusión y Fibroadhesiva 2019Ismael Erazo AstudilloОценок пока нет
- Enfermedad Por Reflujo LaringofaringeoДокумент5 страницEnfermedad Por Reflujo LaringofaringeoDari Salinas SánchezОценок пока нет
- Teorías Del EnvejecimientoДокумент3 страницыTeorías Del EnvejecimientofranklinОценок пока нет
- Tratamiento de Hipoacusia ModeradaДокумент1 страницаTratamiento de Hipoacusia ModeradadabeagboОценок пока нет
- Neurona Motora S e IДокумент35 страницNeurona Motora S e IAlan Alvarez PolinaОценок пока нет
- Neurobiología Del Lenguaje RUBYДокумент18 страницNeurobiología Del Lenguaje RUBYRuBy SánchezОценок пока нет
- Sindrome TopográficosДокумент18 страницSindrome Topográficosbrunocs10Оценок пока нет
- Síndrome de Prader WilliДокумент29 страницSíndrome de Prader WilliEsteban Nicolás Moreno BermedoОценок пока нет
- SPW Prader WillisДокумент5 страницSPW Prader WillisMax OregliaОценок пока нет
- Mamá y PapáДокумент1 страницаMamá y PapáAnggela YáñezОценок пока нет
- Guía Práctica para El Fonema LДокумент1 страницаGuía Práctica para El Fonema LAnggela YáñezОценок пока нет
- Fonema MДокумент4 страницыFonema MAnggela YáñezОценок пока нет
- Guia para Clases Yoga y Meditación InfantilДокумент28 страницGuia para Clases Yoga y Meditación InfantilAnggela Yáñez100% (1)
- Molde EtiquetasДокумент1 страницаMolde EtiquetasAnggela YáñezОценок пока нет
- Y Les Gusta CaminarДокумент2 страницыY Les Gusta CaminarAnggela YáñezОценок пока нет
- Textos Cortos Trabadas RДокумент9 страницTextos Cortos Trabadas RAna Carrasco33% (3)
- Qué Es Un Horario VisualДокумент1 страницаQué Es Un Horario VisualAnggela YáñezОценок пока нет
- Trabalenguas Sinfones RДокумент5 страницTrabalenguas Sinfones RMiriam Grecas100% (2)
- Guia de Aprendizaje I Lenguaje Segundos AñosДокумент10 страницGuia de Aprendizaje I Lenguaje Segundos AñosAnggela YáñezОценок пока нет
- Qué Es Un Horario VisualДокумент1 страницаQué Es Un Horario VisualAnggela YáñezОценок пока нет
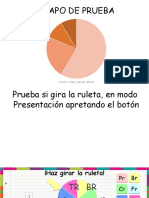
- RuletaДокумент4 страницыRuletaAnggela YáñezОценок пока нет
- Guia para Lograr y Afianzar Difonos Consonanticos Con L 180809173737Документ72 страницыGuia para Lograr y Afianzar Difonos Consonanticos Con L 180809173737Kaathy MardonesОценок пока нет
- Une Los Sonidos R SuaveДокумент1 страницаUne Los Sonidos R SuaveAnggela YáñezОценок пока нет
- Resumen Libro Las BrujasДокумент2 страницыResumen Libro Las BrujasDaniela Venegas Dinamarca73% (86)
- PowerpointДокумент1 страницаPowerpointAnggela YáñezОценок пока нет
- Tarea PolisilabicasДокумент1 страницаTarea PolisilabicasAnggela YáñezОценок пока нет
- Resumen Power PointДокумент3 страницыResumen Power PointAnggela YáñezОценок пока нет
- Inferencias NiñosДокумент5 страницInferencias NiñosAnggela YáñezОценок пока нет
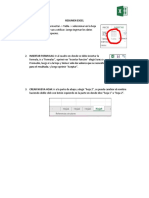
- Resumen ExcelДокумент1 страницаResumen ExcelAnggela YáñezОценок пока нет
- Rol Del FonoДокумент2 страницыRol Del FonoAnggela YáñezОценок пока нет
- Resumen WordДокумент4 страницыResumen WordAnggela YáñezОценок пока нет
- Resumen ExcelДокумент1 страницаResumen ExcelAnggela YáñezОценок пока нет
- CLASE 2 Power PointДокумент1 страницаCLASE 2 Power PointAnggela YáñezОценок пока нет
- Resumen Power PointДокумент3 страницыResumen Power PointAnggela YáñezОценок пока нет
- CLASE 4 Power PointДокумент1 страницаCLASE 4 Power PointAnggela YáñezОценок пока нет
- IdtelДокумент1 страницаIdtelAnggela YáñezОценок пока нет
- CLASE 1 Power PointДокумент1 страницаCLASE 1 Power PointAnggela YáñezОценок пока нет
- CLASE 2 Power PointДокумент1 страницаCLASE 2 Power PointAnggela YáñezОценок пока нет
- IDTELДокумент11 страницIDTELGisel Tapia ArayaОценок пока нет
- Medicina Forense. Guía BásicaДокумент11 страницMedicina Forense. Guía BásicaElizabeth ColínОценок пока нет
- Danyh TrabajoДокумент1 страницаDanyh TrabajogladimiraОценок пока нет
- Médula Espinal - Vías AscendentesДокумент47 страницMédula Espinal - Vías AscendentesNicolás Alejandro Peña Toro90% (21)
- Proceso de RespiraciónДокумент15 страницProceso de RespiraciónLucero BautistaОценок пока нет
- Sistema NerviosoДокумент2 страницыSistema NerviosoThanrry DuarteОценок пока нет
- Inseminacion Artificial en PorcinosДокумент130 страницInseminacion Artificial en PorcinosCarlos GuzmanОценок пока нет
- Casos Clinicos Miembro SuperiorДокумент7 страницCasos Clinicos Miembro SuperiorJonathan RiosОценок пока нет
- Informe de Genética HERENCIA POLIGENICAДокумент16 страницInforme de Genética HERENCIA POLIGENICAAlex Gabriel Soto PintoОценок пока нет
- BacteriasДокумент7 страницBacteriasyorkaОценок пока нет
- Exploración Del Aparato Circulatorio. RUMIANTESДокумент21 страницаExploración Del Aparato Circulatorio. RUMIANTESNery Salinas Colarte71% (7)
- Manual de Tecnicas QuirurgicasДокумент246 страницManual de Tecnicas QuirurgicasTovar Armando100% (5)
- Banco de Preguntas Mediastino y TimoДокумент7 страницBanco de Preguntas Mediastino y TimoCESAR AUGUSTO ARIAS MOLANO0% (1)
- Actividades Evaluativas 2 Adn y Arn y MembranaДокумент5 страницActividades Evaluativas 2 Adn y Arn y MembranaLeidy Huertas BeltranОценок пока нет
- Endocrinología, Metabolismo y Nutrición CTO 3.0Документ168 страницEndocrinología, Metabolismo y Nutrición CTO 3.0Luis YanchapaxiОценок пока нет
- Lista de Pares EmocionalesДокумент7 страницLista de Pares EmocionalesFederico Esaprza100% (1)
- NutricionДокумент22 страницыNutricionHelena RomeroОценок пока нет
- Toxinas VegetalesДокумент7 страницToxinas VegetalesdracyОценок пока нет
- Importancia Del Recurso Humano en La Central de EsterilizaciónДокумент39 страницImportancia Del Recurso Humano en La Central de Esterilizaciónbiomedica.hmuОценок пока нет
- Sentido Del GustoДокумент14 страницSentido Del GustoJazmin RojasОценок пока нет
- Equilibrio Del PotasioДокумент13 страницEquilibrio Del PotasioCamila GonzálezОценок пока нет
- Oxigenoterapia en NiñosДокумент13 страницOxigenoterapia en NiñosAres Amor100% (1)
- Laboratorio N 3 MitosisДокумент2 страницыLaboratorio N 3 MitosisLizondro EG Eva ItzelОценок пока нет
- 8° Evaluación CienciasДокумент4 страницы8° Evaluación CienciasDaniela Ceballos AlarconОценок пока нет
- Masajes ShantalaДокумент21 страницаMasajes ShantalaJaquescb100% (1)
- Guia HipotiroidismoДокумент28 страницGuia HipotiroidismoRocio GuzmanОценок пока нет
- 12 Cosas Comunes Que Dan Miedo Bajo El MicroscopioДокумент23 страницы12 Cosas Comunes Que Dan Miedo Bajo El MicroscopioAnonymous I78Rzn6rSОценок пока нет
- Técnicas de MeditaciónДокумент8 страницTécnicas de Meditaciónfabii BustamanteОценок пока нет
- Aproximacion de La Distribucion Normal A La BinomialДокумент6 страницAproximacion de La Distribucion Normal A La BinomialSofía VillarruelОценок пока нет
- Cámara PulparДокумент3 страницыCámara PulparRonal Mamani Pacheco33% (3)
- Patologías de Oído ExternoДокумент1 страницаPatologías de Oído ExternoFrancisca María López SaavedraОценок пока нет