Вам также может понравиться
- Software Quality Assurance Notes in Urdu Hindi ExplanationДокумент5 страницSoftware Quality Assurance Notes in Urdu Hindi ExplanationAli RazaОценок пока нет
- EMA - Reflection Paper For Laboratories That Perform The Analysis or Evaluation of Clinical Trial SamplesДокумент19 страницEMA - Reflection Paper For Laboratories That Perform The Analysis or Evaluation of Clinical Trial Samplesrpg1973Оценок пока нет
- Computerized Systems Periodic Review: QA Manager - IT ValidationsДокумент4 страницыComputerized Systems Periodic Review: QA Manager - IT ValidationsMaruf RasselОценок пока нет
- Bilgisayarlı SistemlerДокумент14 страницBilgisayarlı Sistemlerttugce29Оценок пока нет
- 5991-8176EN Demystifying Software Validation WhitepaperДокумент7 страниц5991-8176EN Demystifying Software Validation WhitepaperMykolaОценок пока нет
- What Is GAMPДокумент2 страницыWhat Is GAMPsrinivask01Оценок пока нет
- GAMP 5 Categories, V Model, 21 CFR Part 11, EU Annex 11 - AmpleLogicДокумент7 страницGAMP 5 Categories, V Model, 21 CFR Part 11, EU Annex 11 - AmpleLogicArjun TalwakarОценок пока нет
- Using Empower Systemsqt Qualification Tool For Waters Modular HPLC SystemsДокумент8 страницUsing Empower Systemsqt Qualification Tool For Waters Modular HPLC SystemsПетр КрасновОценок пока нет
- Astm-E2500 PDFДокумент2 страницыAstm-E2500 PDFPrashhant KavalleОценок пока нет
- 9 Temperature Mapping Mistakes To AvoidДокумент5 страниц9 Temperature Mapping Mistakes To AvoidReema IshaqОценок пока нет
- Eres Annex 11 Eu GMP SiemensДокумент30 страницEres Annex 11 Eu GMP SiemenshuykhiemОценок пока нет
- Minitab-2003-Software ValidationДокумент11 страницMinitab-2003-Software ValidationGonzalo_Rojas_VerenzОценок пока нет
- Pharmaceutical Quality Audits: A ReviewДокумент9 страницPharmaceutical Quality Audits: A ReviewHema PepakayalaОценок пока нет
- Selecting, Implementing and Using FDA Compliance Software SolutionsДокумент29 страницSelecting, Implementing and Using FDA Compliance Software SolutionsSireeshaОценок пока нет
- VAL 135 Risk Assessment For Computer Validation Systems Sample - SandraДокумент3 страницыVAL 135 Risk Assessment For Computer Validation Systems Sample - SandraSandra Silva100% (1)
- Risk Profile of The SystemДокумент7 страницRisk Profile of The SystemAbhijeetОценок пока нет
- QC Supervisor or QC Manager or QA Supervisor or QA Manager or ReДокумент2 страницыQC Supervisor or QC Manager or QA Supervisor or QA Manager or Reapi-77380607Оценок пока нет
- Lecture 9 - QAQC PDFДокумент37 страницLecture 9 - QAQC PDFTMTОценок пока нет
- Data and Database Integrity TestingДокумент3 страницыData and Database Integrity TestingMounir Ben MohamedОценок пока нет
- Presentation #1 - Effective InvestigationsДокумент26 страницPresentation #1 - Effective InvestigationsgoodgodnoОценок пока нет
- A History of The OOS ProblemДокумент5 страницA History of The OOS ProblemmcyqcbsacОценок пока нет
- Urs For Erp Ex 2Документ37 страницUrs For Erp Ex 2AbhijeetОценок пока нет
- Hazop PharmaДокумент7 страницHazop PharmaismailayarОценок пока нет
- Validation Master Plan (VMP)Документ11 страницValidation Master Plan (VMP)Gonzalo MazaОценок пока нет
- Control Strategy Case StudiesДокумент25 страницControl Strategy Case StudiesNarongchai PongpanОценок пока нет
- 21CFR11 Assessment FAQ Metler Toledo STAREДокумент51 страница21CFR11 Assessment FAQ Metler Toledo STAREfurqan.malikОценок пока нет
- Lab Notebook Presentation - 8-12-2016Документ44 страницыLab Notebook Presentation - 8-12-2016Muhammad ALi SoomroОценок пока нет
- Aada: Aao: Ade: Adme: Adi: Adr: Adrs: Agdufa: Ahu: Alcoa: Alcoa PlusДокумент10 страницAada: Aao: Ade: Adme: Adi: Adr: Adrs: Agdufa: Ahu: Alcoa: Alcoa PlusjhansiОценок пока нет
- GDL GIQAR GCP Gamp Guideline MilanoДокумент77 страницGDL GIQAR GCP Gamp Guideline Milanocristhianjdv50% (2)
- Computer System Validation in Pharmaceutical IndustryДокумент9 страницComputer System Validation in Pharmaceutical IndustryUmair HussainОценок пока нет
- Workflows For Quality Risk Management-2Документ24 страницыWorkflows For Quality Risk Management-2sarada jenaОценок пока нет
- ECA Computerised System Validation GAMP 5 ApproachДокумент6 страницECA Computerised System Validation GAMP 5 ApproachHanan NoussaОценок пока нет
- GAMP FR PP20201001 001 v0100EN PDFДокумент5 страницGAMP FR PP20201001 001 v0100EN PDFHarish GopinathОценок пока нет
- FDA Warning Letter For Inadequate Batch Record ReviewДокумент1 страницаFDA Warning Letter For Inadequate Batch Record ReviewMina Maher MikhailОценок пока нет
- FMEA Risk Analysis ExampleДокумент1 страницаFMEA Risk Analysis ExampleDavid SigalinggingОценок пока нет
- Deviations and Non Conformances SOP PDFДокумент1 страницаDeviations and Non Conformances SOP PDFAlinaОценок пока нет
- LIMS For Lasers 2015 User ManualДокумент188 страницLIMS For Lasers 2015 User Manualcharles_0814Оценок пока нет
- Cleaning Validation Boot CampДокумент7 страницCleaning Validation Boot Campramin_47Оценок пока нет
- Computer System Validation Complete Guide 1710042264Документ105 страницComputer System Validation Complete Guide 1710042264My TalentОценок пока нет
- Aplicabilidad 21CFR11Документ6 страницAplicabilidad 21CFR11Aydee RojasОценок пока нет
- CSV 2 Gamp5Документ14 страницCSV 2 Gamp5manoj DhamneОценок пока нет
- Capa-UpДокумент22 страницыCapa-UpChristinaОценок пока нет
- Change ControlДокумент2 страницыChange Controlbhavik_meghaniОценок пока нет
- Analytical Equipment Lifecycle ManagementДокумент24 страницыAnalytical Equipment Lifecycle Managementtvijayak41Оценок пока нет
- Malvern Access Configurator (Mac) User Guide: MAN0602-01-EN-00 July 2017Документ50 страницMalvern Access Configurator (Mac) User Guide: MAN0602-01-EN-00 July 2017Pentesh NingaramainaОценок пока нет
- Guidelines For The Development and Validation of Spreadsheets PDFДокумент21 страницаGuidelines For The Development and Validation of Spreadsheets PDFraj kumar chaudharyОценок пока нет
- Computer System Validation: Embedding Quality Into Automated ProcessesДокумент29 страницComputer System Validation: Embedding Quality Into Automated ProcessesMohamed KamalОценок пока нет
- Cchem Applicant RequirementsДокумент11 страницCchem Applicant RequirementsbukkysuccessОценок пока нет
- GMP Advisor: The GMP Questions & Answers GuideДокумент152 страницыGMP Advisor: The GMP Questions & Answers GuidedikshaОценок пока нет
- Basic Principles of GMP Basic Principles of GMPДокумент33 страницыBasic Principles of GMP Basic Principles of GMPTahir KhanОценок пока нет
- A-Mab Case Study Version 2-1Документ278 страницA-Mab Case Study Version 2-1Davion Stewart100% (1)
- Medical Erp Buyer S GuideДокумент16 страницMedical Erp Buyer S GuideNikhil PrasannaОценок пока нет
- Overview of Validation Documents and ProjectsДокумент5 страницOverview of Validation Documents and ProjectsMD Fahad MiajiОценок пока нет
- Root Causes and CAPAДокумент27 страницRoot Causes and CAPAsanjeev kumarОценок пока нет
- User Requirement SpecificationДокумент78 страницUser Requirement Specificationgigichung1Оценок пока нет
- Data Integrity and Compliance: A Primer for Medical Product ManufacturersОт EverandData Integrity and Compliance: A Primer for Medical Product ManufacturersОценок пока нет
- A Beginner S Guide To Effective Programming: Mahindra Satyam Learning WorldДокумент265 страницA Beginner S Guide To Effective Programming: Mahindra Satyam Learning WorldIshmo KueedОценок пока нет
- Easy Math CalculationДокумент4 страницыEasy Math CalculationJayОценок пока нет
- CAN Cable ConnectionДокумент1 страницаCAN Cable Connectionsatish734Оценок пока нет
- Maxon RE40 Graphite Brushes 150wattДокумент1 страницаMaxon RE40 Graphite Brushes 150wattElectromateОценок пока нет
- Brochure of Broken Bag Detector PDFДокумент4 страницыBrochure of Broken Bag Detector PDFsatish734Оценок пока нет
- Pvi BaseДокумент176 страницPvi Basesatish734Оценок пока нет
- Extrusion Spheronization - A ReviewДокумент5 страницExtrusion Spheronization - A Reviewsatish734Оценок пока нет
- How To Make Symbols With KeyboardДокумент1 страницаHow To Make Symbols With KeyboardMayzatt MaisyaОценок пока нет
- Euroclean Pro LeafletДокумент2 страницыEuroclean Pro Leafletsatish734Оценок пока нет
- CGL Make Flame Proof MotorsДокумент19 страницCGL Make Flame Proof MotorsKishore KrishnaОценок пока нет
- 108 Sharana GhoshamДокумент1 страница108 Sharana Ghoshamsatish734Оценок пока нет
- Abb Acs355 CatalogДокумент24 страницыAbb Acs355 Catalogsatish734Оценок пока нет
- Policy Holder's Manual - Private Car Package: Bajaj Allianz General Insurance Company LTDДокумент33 страницыPolicy Holder's Manual - Private Car Package: Bajaj Allianz General Insurance Company LTDCharlie SinghОценок пока нет
- Abb Acs355 CatalogДокумент24 страницыAbb Acs355 Catalogsatish734Оценок пока нет
- Kerla S Kranti RouteДокумент1 страницаKerla S Kranti Routesatish734Оценок пока нет
- Granulation With Rapid Mixer Granulator (RMG) - A Review - Pharma InfoДокумент42 страницыGranulation With Rapid Mixer Granulator (RMG) - A Review - Pharma Infosatish734Оценок пока нет
- Tm450 Acopos Control Concept and AdjustmentДокумент51 страницаTm450 Acopos Control Concept and Adjustmentsatish734Оценок пока нет
- Abb Acs355 CatalogДокумент24 страницыAbb Acs355 Catalogsatish734Оценок пока нет
- XTS Beckhoff eДокумент32 страницыXTS Beckhoff esatish734Оценок пока нет
- Cricket ScoresheetДокумент1 страницаCricket ScoresheetTagenarine SinghОценок пока нет
- GMPДокумент4 страницыGMPsatish734Оценок пока нет
- Cricket ScoresheetДокумент1 страницаCricket ScoresheetTagenarine SinghОценок пока нет
- Wagon R BrochureДокумент11 страницWagon R Brochuremuthu189Оценок пока нет
- Practical Electronics 1968 10 S OCRДокумент84 страницыPractical Electronics 1968 10 S OCRCarlos SoaresОценок пока нет
- Compact and Reliable Switching - Smaller, More Efficient and Simpler Gas-Insulated Switchgear (GIS)Документ6 страницCompact and Reliable Switching - Smaller, More Efficient and Simpler Gas-Insulated Switchgear (GIS)qxzyОценок пока нет
- GM 302 - Update - 10-2004Документ26 страницGM 302 - Update - 10-2004Naser JahangiriОценок пока нет
- Test AND Measurement: Eagle PhotonicsДокумент90 страницTest AND Measurement: Eagle PhotonicsPankaj SharmaОценок пока нет
- MU-MIMO in LTEДокумент13 страницMU-MIMO in LTECM_123Оценок пока нет
- RAMS Knowledge SharingДокумент29 страницRAMS Knowledge SharingJAYОценок пока нет
- Ryan Donnelly - Artificial Intelligence in GamingДокумент12 страницRyan Donnelly - Artificial Intelligence in GamingJavier Pardo MuñozОценок пока нет
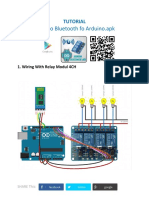
- Arduino Bluetooth Ralay 4chДокумент5 страницArduino Bluetooth Ralay 4chRaul Lara RochaОценок пока нет
- EVSДокумент7 страницEVSsubhas9804009247Оценок пока нет
- Ivd Symbols FinalДокумент14 страницIvd Symbols FinalDennis ChenОценок пока нет
- WWW - Manaresults.Co - In: Set No. 1Документ1 страницаWWW - Manaresults.Co - In: Set No. 1My Technical videosОценок пока нет
- Yamaha Acoustic GuitarsДокумент18 страницYamaha Acoustic Guitarsrusf123100% (5)
- FM 5-102 - CountermobilityДокумент220 страницFM 5-102 - CountermobilitySurvivIt100% (2)
- Ram 100Документ2 страницыRam 100MAT-LIONОценок пока нет
- Perdele Economic B 2VVДокумент4 страницыPerdele Economic B 2VVakitainupufОценок пока нет
- Class 6 Ioel 2017Документ8 страницClass 6 Ioel 2017A GuptaОценок пока нет
- HRSG Design and Operation On Unit Reliability and Remaining LifeДокумент74 страницыHRSG Design and Operation On Unit Reliability and Remaining LifeNisal PereraОценок пока нет
- Ogm 25Документ1 страницаOgm 25azimsabudinОценок пока нет
- HR AuditДокумент5 страницHR AuditshanumanuranuОценок пока нет
- 6.9. How To Send SSL-Encrypted EmailДокумент3 страницы6.9. How To Send SSL-Encrypted EmailJoxОценок пока нет
- Trafo Manual ABBДокумент104 страницыTrafo Manual ABBMarcos SebastianОценок пока нет
- System Development Process: The Incremental Commitment ModelДокумент20 страницSystem Development Process: The Incremental Commitment Modeloctopus2011Оценок пока нет
- KW Tedder LeafletДокумент32 страницыKW Tedder Leafletinfo4826Оценок пока нет
- India International Centre India International Centre QuarterlyДокумент15 страницIndia International Centre India International Centre QuarterlySruti UОценок пока нет
- Pressostato SUCO - 0159Документ3 страницыPressostato SUCO - 0159Hugo Lemos ArthusoОценок пока нет
- Adjustment of Conditions: Contents: Procedure Adjustment Methods Country-Specific Methods in Austria and SwitzerlandДокумент41 страницаAdjustment of Conditions: Contents: Procedure Adjustment Methods Country-Specific Methods in Austria and Switzerlandtushar2001Оценок пока нет
- DC DC CONVERT DS en 12Документ2 страницыDC DC CONVERT DS en 12carlos G7Оценок пока нет
- RoundingДокумент65 страницRoundingSourav Kumar100% (1)
- PDH DFE1000 BrochureДокумент2 страницыPDH DFE1000 Brochuremajdi1985Оценок пока нет
- 17 - Defining Service Level Agreement (SLA) For E-Gov ProjectsДокумент9 страниц17 - Defining Service Level Agreement (SLA) For E-Gov ProjectsdevОценок пока нет