Вам также может понравиться
- Dengue Casos Clinicos Pra Revalida BrasilДокумент170 страницDengue Casos Clinicos Pra Revalida BrasilAlejandro Josue Zurita Chuca100% (1)
- Diagnóstico Das Hepatites ViraisДокумент93 страницыDiagnóstico Das Hepatites ViraisAlejandro Josue Zurita ChucaОценок пока нет
- MapaMentalHepatiteC 1528909247700Документ1 страницаMapaMentalHepatiteC 1528909247700Alejandro Josue Zurita ChucaОценок пока нет
- 9MapamentalLqAmnitico 1531152943659 PDFДокумент1 страница9MapamentalLqAmnitico 1531152943659 PDFAlejandro Josue Zurita ChucaОценок пока нет
- MapaPrnatal 1526062872363Документ1 страницаMapaPrnatal 1526062872363Alejandro Josue Zurita ChucaОценок пока нет
- Casos Clinicos Das Hepatites - Sus RevalidaДокумент100 страницCasos Clinicos Das Hepatites - Sus RevalidaAlejandro Josue Zurita Chuca100% (1)
- AclsДокумент2 страницыAclsAlejandro Josue Zurita ChucaОценок пока нет
- MapamentalEsteatohepatitenoalcolica 1550108612117Документ1 страницаMapamentalEsteatohepatitenoalcolica 1550108612117Alejandro Josue Zurita Chuca100% (1)
- Colite Pseudomenbranosa: Clostridium DifficileДокумент1 страницаColite Pseudomenbranosa: Clostridium DifficileAlejandro Josue Zurita ChucaОценок пока нет
- MapamentalAscite 1550108504871Документ1 страницаMapamentalAscite 1550108504871Alejandro Josue Zurita Chuca100% (1)
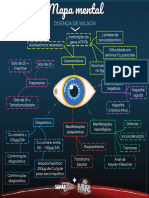
- MapamentalDoenadeWilson 1552674544408Документ1 страницаMapamentalDoenadeWilson 1552674544408Alejandro Josue Zurita ChucaОценок пока нет
- MapamentalCncerHeptico1546466666106 1554402321672Документ1 страницаMapamentalCncerHeptico1546466666106 1554402321672Alejandro Josue Zurita ChucaОценок пока нет
- MapamentalDisfagia 1550108972595Документ1 страницаMapamentalDisfagia 1550108972595Alejandro Josue Zurita ChucaОценок пока нет
- Diarreia 1527704419280Документ1 страницаDiarreia 1527704419280Alejandro Josue Zurita ChucaОценок пока нет
- MapaMentalIntoxicaesExgenasAgudas 1550108460185Документ1 страницаMapaMentalIntoxicaesExgenasAgudas 1550108460185Alejandro Josue Zurita ChucaОценок пока нет
- Dreg 1526922421924Документ1 страницаDreg 1526922421924Alejandro Josue Zurita ChucaОценок пока нет
- Hipertireoidismo: CausasДокумент1 страницаHipertireoidismo: CausasAlejandro Josue Zurita ChucaОценок пока нет
- MapamentalDislipidemias 1550108429837Документ1 страницаMapamentalDislipidemias 1550108429837Alejandro Josue Zurita Chuca100% (1)
- MapaMentalChoque 1534807809941Документ1 страницаMapaMentalChoque 1534807809941Alejandro Josue Zurita ChucaОценок пока нет
- Infecções Do Esôfago: Herpes SimplexДокумент1 страницаInfecções Do Esôfago: Herpes SimplexAlejandro Josue Zurita ChucaОценок пока нет
- Cetoacidose DiabéticaДокумент1 страницаCetoacidose DiabéticaAlejandro Josue Zurita ChucaОценок пока нет
- Abdome Agudo VascularДокумент1 страницаAbdome Agudo VascularAlejandro Josue Zurita ChucaОценок пока нет
- Reanimação NeonatalДокумент1 страницаReanimação NeonatalAlejandro Josue Zurita Chuca100% (1)
- MapamentalChoquenacriana 1538150099383Документ1 страницаMapamentalChoquenacriana 1538150099383Alejandro Josue Zurita ChucaОценок пока нет
- DoremabdomeSuperior 1533780729460Документ1 страницаDoremabdomeSuperior 1533780729460Alejandro Josue Zurita ChucaОценок пока нет
- Dispneiamapamental 1533922474206Документ1 страницаDispneiamapamental 1533922474206Alejandro Josue Zurita ChucaОценок пока нет
- EmergnciasPsiquitricas 1533780445804Документ1 страницаEmergnciasPsiquitricas 1533780445804Alejandro Josue Zurita ChucaОценок пока нет
- Diagnósticos Diferenciais de Dor TorácicaДокумент1 страницаDiagnósticos Diferenciais de Dor TorácicaAlejandro Josue Zurita ChucaОценок пока нет
- Doremabdomeinferior 1534806721525Документ1 страницаDoremabdomeinferior 1534806721525Alejandro Josue Zurita ChucaОценок пока нет
- Crianagrave 1533780425367Документ1 страницаCrianagrave 1533780425367Alejandro Josue Zurita ChucaОценок пока нет
- Resumo Do Curso Modo BudaДокумент3 страницыResumo Do Curso Modo BudaRodrigo RiosОценок пока нет
- Aula 6 - AerossóisДокумент24 страницыAula 6 - AerossóisJoab Ana Menezes100% (2)
- (Thompson, 7 Ed) Cap. 16 - Genômica e Genética Do CâncerДокумент6 страниц(Thompson, 7 Ed) Cap. 16 - Genômica e Genética Do CâncerDenis NascimentoОценок пока нет
- n65 - G - Prolapso Dos Orgaos Pelvicos 2Документ16 страницn65 - G - Prolapso Dos Orgaos Pelvicos 2Fernanda KochiОценок пока нет
- Edital PSS 2024 2Документ18 страницEdital PSS 2024 2naylma.santos.2527Оценок пока нет
- Assistente FinanceiroДокумент3 страницыAssistente FinanceiroRENATAОценок пока нет
- Relatório Visita Ao Caps AdДокумент4 страницыRelatório Visita Ao Caps AdGabriela NaboznyОценок пока нет
- Laudo de Avaliação de Emissão de RuidoДокумент5 страницLaudo de Avaliação de Emissão de RuidoJesus PassosОценок пока нет
- Infarmed - Farmacovigilancia PDFДокумент580 страницInfarmed - Farmacovigilancia PDFPatrícia BarrosОценок пока нет
- Anexo B It 42Документ3 страницыAnexo B It 42Adalberto LimaОценок пока нет
- Prova Medicina Legal Matutino B2 2.º 2020Документ5 страницProva Medicina Legal Matutino B2 2.º 2020Alex RezendeОценок пока нет
- Prova Bioquimica EnfermagemДокумент3 страницыProva Bioquimica EnfermagemBruno De Andrade Pires100% (1)
- Catalogo Ofertas Marzo 2023 (Al 07-03)Документ655 страницCatalogo Ofertas Marzo 2023 (Al 07-03)nicoleОценок пока нет
- Treinamento AdmissionalДокумент2 страницыTreinamento Admissionalivanzinho DiasОценок пока нет
- Cuidados Com o Ambiente D EtrabalhoДокумент4 страницыCuidados Com o Ambiente D EtrabalhoAlda KoglinОценок пока нет
- Apontamentos Sistema DigestivoДокумент6 страницApontamentos Sistema DigestivoEster Machado DalilaОценок пока нет
- A Gestão Do Tempo É Muito Importante para Um AlunoДокумент5 страницA Gestão Do Tempo É Muito Importante para Um AlunoKalitangui NunesОценок пока нет
- POP - 05 ArmazenamentoДокумент5 страницPOP - 05 ArmazenamentoWALBER AraujoОценок пока нет
- A Tutela Dos Trabalhadores Portadores de HIV e SIDA Uma Análise À Luz Do Ordenamento Jurídico-Laboral Moçambicano - Admira Maria Raúl Sábado - 2015Документ52 страницыA Tutela Dos Trabalhadores Portadores de HIV e SIDA Uma Análise À Luz Do Ordenamento Jurídico-Laboral Moçambicano - Admira Maria Raúl Sábado - 2015Elísio NhamicocheОценок пока нет
- Aquisição, Recebimento e Armazenamento: Reponsabilidade FarmacêuticoДокумент6 страницAquisição, Recebimento e Armazenamento: Reponsabilidade FarmacêuticoSOS CARMOОценок пока нет
- Formação Tigresa de JadeДокумент18 страницFormação Tigresa de JadeLola GillianОценок пока нет
- Pessoal Vjxxqc3y.wagДокумент176 страницPessoal Vjxxqc3y.waglmarxОценок пока нет
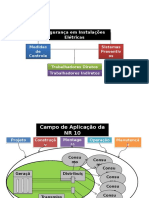
- NR 10 EsquematizadaДокумент10 страницNR 10 EsquematizadaArthur Danc100% (1)
- Habilitações BiomedicinaДокумент7 страницHabilitações BiomedicinaÉrika BrandãoОценок пока нет
- Prova Objetiva - Cod. 08 - Nutricionista - Tipo 2Документ14 страницProva Objetiva - Cod. 08 - Nutricionista - Tipo 2Patricia de MoraisОценок пока нет
- Guia Rolo CompactadorДокумент1 страницаGuia Rolo CompactadorCleibe Vaz MachadoОценок пока нет
- Form Obj 0Документ18 страницForm Obj 0anderson640luiz82Оценок пока нет
- Psicologo EmpreendedorДокумент33 страницыPsicologo EmpreendedorFranciscoWalisson100% (1)
- Modelo Slide Jornada EnfermagemДокумент5 страницModelo Slide Jornada EnfermagemLetícia MachadoОценок пока нет
- Projeto Visão Nas EscolasДокумент43 страницыProjeto Visão Nas EscolasFrancisco XavierОценок пока нет