Вам также может понравиться
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5782)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (265)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2219)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (119)
- Alex Kaivarainen - New Hierarchic Theory of Water and Its Role in Biosystems: The Quantum Psi ProblemДокумент54 страницыAlex Kaivarainen - New Hierarchic Theory of Water and Its Role in Biosystems: The Quantum Psi ProblemJoaokaaОценок пока нет
- COVID-19 Salivary Signature: Diagnostic and Research OpportunitiesДокумент6 страницCOVID-19 Salivary Signature: Diagnostic and Research OpportunitiesKalyan KarumanchiОценок пока нет
- Patent US20120251502 - Human Ebola Virus Species and Compositions and Methods Thereof - Google PatenteДокумент32 страницыPatent US20120251502 - Human Ebola Virus Species and Compositions and Methods Thereof - Google PatenteSimona von BrownОценок пока нет
- # 176 Evaluation of Malignant Ascites: Fast Facts and Concepts #176Документ21 страница# 176 Evaluation of Malignant Ascites: Fast Facts and Concepts #176Abdul QuyyumОценок пока нет
- Dr. G. M. Taori: Curriculum VitaeДокумент12 страницDr. G. M. Taori: Curriculum Vitaesrajan sahuОценок пока нет
- Multiple Myeloma: NCCN Guidelines For PatientsДокумент78 страницMultiple Myeloma: NCCN Guidelines For PatientsAchmad Farodisi AfnaniОценок пока нет
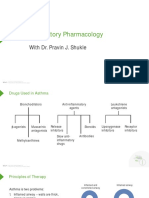
- Respiratory Pharmacology: With Dr. Pravin J. ShukleДокумент28 страницRespiratory Pharmacology: With Dr. Pravin J. ShukleKaish DahiyaОценок пока нет
- Expanded Immunization Program EpiДокумент22 страницыExpanded Immunization Program EpiGirome BairaОценок пока нет
- Clinical ImmunologyДокумент414 страницClinical ImmunologyCaio LimaОценок пока нет
- Rabies SupplementalsДокумент6 страницRabies SupplementalsJoher Bolante MendezОценок пока нет
- RootДокумент119 страницRootเทพนิมิตร สมภักดีОценок пока нет
- Vetscan ObjectivesДокумент395 страницVetscan ObjectivesDeep PatelОценок пока нет
- AiG - Genetics - Human GenomeДокумент53 страницыAiG - Genetics - Human Genomest iОценок пока нет
- Form 2 Chapter 4 Human HealthДокумент30 страницForm 2 Chapter 4 Human HealthAmer MalekОценок пока нет
- Live All Questions Final2021Документ50 страницLive All Questions Final2021Hab AnneОценок пока нет
- Brucellosis Is A Disease Caused by Bacteria in The Genus BrucellaДокумент28 страницBrucellosis Is A Disease Caused by Bacteria in The Genus BrucellaAhmed J AlhindaweОценок пока нет
- Nej MR A 1206793Документ13 страницNej MR A 1206793David LeónОценок пока нет
- Anatomy and Physiology Related To Multiple Myelom1Документ15 страницAnatomy and Physiology Related To Multiple Myelom1Diane Kate Tobias Magno100% (1)
- ATP Blog - Germanium by - Dr. Sandra GoodmanДокумент101 страницаATP Blog - Germanium by - Dr. Sandra GoodmanatpfacebookОценок пока нет
- Immunohema Midterm ReviewerДокумент21 страницаImmunohema Midterm ReviewerKJDTolentinoОценок пока нет
- Biotechnology A Problem Approach PDFДокумент247 страницBiotechnology A Problem Approach PDFChandu P Lal0% (1)
- New Microsoft Word DocumentДокумент7 страницNew Microsoft Word DocumentDocAxi Maximo Jr AxibalОценок пока нет
- Innate and Adaptive Immunity ExplainedДокумент5 страницInnate and Adaptive Immunity ExplainedKebbewar KhaledОценок пока нет
- Vidya Sagar PhsyologyДокумент55 страницVidya Sagar PhsyologyHaris QurashiОценок пока нет
- Che514a IntroductionДокумент30 страницChe514a IntroductionGenevive S. de VeraОценок пока нет
- Biological Products and Their Impact on Human Cells and TissuesДокумент28 страницBiological Products and Their Impact on Human Cells and TissuesShintia Surya PutriОценок пока нет
- 2010-11 partA-F eДокумент41 страница2010-11 partA-F eAltunc Suleyman GezerlerОценок пока нет
- L1a AntibodiesДокумент37 страницL1a Antibodiesashutoshrath209Оценок пока нет
- 1.2 Immunity Response TransДокумент10 страниц1.2 Immunity Response TransJoshua SaanОценок пока нет
- ANSWER SCHEME CHAPTER 1Документ24 страницыANSWER SCHEME CHAPTER 1Chen ShyanОценок пока нет