Вам также может понравиться
- Ejercicio 7Документ7 страницEjercicio 7Leydi Torcagua Huamani100% (1)
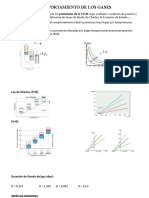
- Leyes de los gases ideales y ecuación de estado PV=nRTДокумент9 страницLeyes de los gases ideales y ecuación de estado PV=nRTRAUL FERREYRA GARCIAОценок пока нет
- Teoría Del Gas Real: La Relación Exacta Para Gases RealesОт EverandTeoría Del Gas Real: La Relación Exacta Para Gases RealesОценок пока нет
- NVF 0928 2008Документ162 страницыNVF 0928 2008MIGUEL RODRIGUEZ INGENIERO CIVIL ULA VEОценок пока нет
- Ejercicios Resueltos Balances de EnergiaДокумент9 страницEjercicios Resueltos Balances de EnergiaJUAN DIEGO VILLAMIL SIERRA100% (1)
- Principio de Bernoulli ModificadoДокумент19 страницPrincipio de Bernoulli ModificadoladyОценок пока нет
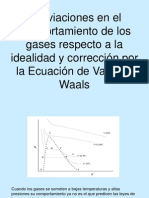
- Gases reales y ecuación de Van der WaalsДокумент9 страницGases reales y ecuación de Van der WaalsCamila PachecoОценок пока нет
- Quimica General IДокумент18 страницQuimica General ISofía Alvarez HernandezОценок пока нет
- Ecuaciones gases realesДокумент13 страницEcuaciones gases realesCristian Ccaso MamaniОценок пока нет
- Ecuacion de Van Der WaalsДокумент3 страницыEcuacion de Van Der WaalsviridianaОценок пока нет
- Comportamiento de Los GasesДокумент25 страницComportamiento de Los GasesElsa Marzana SanizoОценок пока нет
- Comportamiento de los gasesДокумент25 страницComportamiento de los gasesERIKA LOZADA RUSSELОценок пока нет
- Cap 1 Gases Reales. Parte 1 Gases HúmedosДокумент41 страницаCap 1 Gases Reales. Parte 1 Gases HúmedosJhamil CondeОценок пока нет
- La Ecuación de ClapeyronДокумент8 страницLa Ecuación de ClapeyronHg RosmeriОценок пока нет
- La Ecuación de ClapeyronДокумент8 страницLa Ecuación de ClapeyronHg RosmeriОценок пока нет
- Termo 3Документ36 страницTermo 3José Emilio GuardiaОценок пока нет
- Gases Reales PresentacionДокумент48 страницGases Reales PresentacionEdwinОценок пока нет
- Ecuaciones de estado para gases ideales y realesДокумент63 страницыEcuaciones de estado para gases ideales y realesCallejas Palominos Eduardo0% (1)
- 01 - Química - Leyes de Los GasesДокумент7 страниц01 - Química - Leyes de Los GasesCarlos SuárezОценок пока нет
- Dilatación TérmicaДокумент10 страницDilatación TérmicaLeonardo WalterОценок пока нет
- Clase II, Gases Reales, Ecuacion de Van Der WaalsДокумент13 страницClase II, Gases Reales, Ecuacion de Van Der WaalsDoubtxxAnxiety0% (1)
- Termodinámica de gases: Leyes y ecuacionesДокумент34 страницыTermodinámica de gases: Leyes y ecuacionespatriarca28Оценок пока нет
- Guia de Lab - Ley de CharlesДокумент7 страницGuia de Lab - Ley de CharlesMaria JoseОценок пока нет
- Quimica General Gases Ejercicios Tipo ResueltosДокумент12 страницQuimica General Gases Ejercicios Tipo ResueltosCamilo AlbornozОценок пока нет
- Lab N°3 (Van Der Waals)Документ10 страницLab N°3 (Van Der Waals)Luis Dani Torres GuerraОценок пока нет
- Practica 1 Quimica AplicadaДокумент10 страницPractica 1 Quimica AplicadaAlfredoОценок пока нет
- Clase 1 2019-1 Gases IDEALES-REALESДокумент39 страницClase 1 2019-1 Gases IDEALES-REALESAlberto perez william100% (1)
- Clase 2 Gases RealesДокумент74 страницыClase 2 Gases RealesPETER ANDRES JACOBO ABURTOОценок пока нет
- Leyes de Los GasesДокумент8 страницLeyes de Los GasesEsteban Andres ZabalaОценок пока нет
- TEMA VIII Estado GaseosoДокумент32 страницыTEMA VIII Estado GaseosoGREDY JOSEPH SULCA MARTINEZОценок пока нет
- Clase 5Документ34 страницыClase 5Miguel AngelОценок пока нет
- Gases Reales y Ecuacion Van Der WaalsДокумент12 страницGases Reales y Ecuacion Van Der WaalsLiliana Manzano100% (1)
- Ecuación de Gases RealesДокумент9 страницEcuación de Gases RealesAli DíazОценок пока нет
- Ecuación Van der WaalsДокумент5 страницEcuación Van der WaalsCarlos CarhuayoОценок пока нет
- MÓDULO 4 - GasesДокумент32 страницыMÓDULO 4 - Gasesjose.torres1Оценок пока нет
- Ley de Los Gases Trabajo FinalДокумент9 страницLey de Los Gases Trabajo FinalDaya MirandaОценок пока нет
- Fisico Quimica - Ramirez Castillo Jorge MiguelДокумент40 страницFisico Quimica - Ramirez Castillo Jorge MiguelMiguel CastilloОценок пока нет
- T1 2008 4 Sistemas de Una FaseДокумент29 страницT1 2008 4 Sistemas de Una FaseEduardoLalo MamaniОценок пока нет
- EdE cúbicasДокумент26 страницEdE cúbicasJulio DuarteОценок пока нет
- Gases RealesДокумент35 страницGases RealesMarco Cisneros,Yheferson Maza,Diego Díaz,Jairo Giohayro,David Samamé,Gino Cuadros,Ricardo Trinidad100% (1)
- IntroIQ Clase 17 y 18Документ27 страницIntroIQ Clase 17 y 18AR QuímicaОценок пока нет
- Gases RealesДокумент30 страницGases RealesANGELES NIKOLL FLORES FLORESОценок пока нет
- Leyes de los gases idealesДокумент11 страницLeyes de los gases idealesEspinoza LuisОценок пока нет
- Ec. Van Der WaalsДокумент3 страницыEc. Van Der WaalsItzelCariОценок пока нет
- CAP 1 GASES REALES. Parte 1 Leyes Empíricas Del Gas IdealДокумент20 страницCAP 1 GASES REALES. Parte 1 Leyes Empíricas Del Gas IdealDaniel FeymanОценок пока нет
- 4 - Leyes de Los Gases 2009-1Документ32 страницы4 - Leyes de Los Gases 2009-1Alison RoaОценок пока нет
- Investigacion 2Документ3 страницыInvestigacion 2DiegoAndrèsOrtegaPeraltaОценок пока нет
- Gases y Soluciones RealesДокумент20 страницGases y Soluciones RealesEliza RicothОценок пока нет
- Ecuaciones de Estado para Gases RealesДокумент25 страницEcuaciones de Estado para Gases RealesKEVIN ARNOLDO PEREZ RIVASОценок пока нет
- Ley de CharlesДокумент4 страницыLey de Charlesjoaquin arredondoОценок пока нет
- Instituto Tecnológico Nacional de México Campus Villahermosa Alumno: Catedratica: Asignatura: Carrera: Semestre: FechaДокумент23 страницыInstituto Tecnológico Nacional de México Campus Villahermosa Alumno: Catedratica: Asignatura: Carrera: Semestre: FechaEduardo NiñoОценок пока нет
- Gases Reales - Uso de Ecuaciones de Estado PDFДокумент6 страницGases Reales - Uso de Ecuaciones de Estado PDFGeraldine D. FernándezОценок пока нет
- Termodinámica gases ideales coeficiente expansiónДокумент18 страницTermodinámica gases ideales coeficiente expansiónJesús CubasОценок пока нет
- Energía interna, entalpía y ecuaciones de estado para gases idealesДокумент14 страницEnergía interna, entalpía y ecuaciones de estado para gases idealesAndres AndradeОценок пока нет
- Leyes de Los Gases 2023-IДокумент44 страницыLeyes de Los Gases 2023-IdanielОценок пока нет
- 4.2 Ejercicios de Trabajoy CalorДокумент66 страниц4.2 Ejercicios de Trabajoy CalorBruno CastilloОценок пока нет
- Term Odin A MicaДокумент21 страницаTerm Odin A MicaNel VelaОценок пока нет
- Guia de Laboratorio 10. GasesДокумент12 страницGuia de Laboratorio 10. GasesNeyira PérezОценок пока нет
- Presentacion 1 GasesДокумент62 страницыPresentacion 1 GasesUlises ChavezОценок пока нет
- Ingeniería química. Soluciones a los problemas del tomo IОт EverandIngeniería química. Soluciones a los problemas del tomo IОценок пока нет
- Documento Sin TítuloДокумент5 страницDocumento Sin TítuloLeydi Torcagua HuamaniОценок пока нет
- FitoremediaciónДокумент14 страницFitoremediaciónLeydi Torcagua HuamaniОценок пока нет
- Tabulacion de Encuesta Al Restaurante "Doña Peta": ¿ Mi Jefe Establece Objetivos Realistas?Документ2 страницыTabulacion de Encuesta Al Restaurante "Doña Peta": ¿ Mi Jefe Establece Objetivos Realistas?Leydi Torcagua HuamaniОценок пока нет
- Grupo 1Документ2 страницыGrupo 1Leydi Torcagua HuamaniОценок пока нет
- LegiДокумент8 страницLegiLeydi Torcagua HuamaniОценок пока нет
- Coordenadas Utm Kristofeo XDДокумент15 страницCoordenadas Utm Kristofeo XDLeydi Torcagua HuamaniОценок пока нет
- Qué Vamos A Indagar Acerca de Los ContinentesДокумент1 страницаQué Vamos A Indagar Acerca de Los ContinentesLeydi Torcagua HuamaniОценок пока нет
- 1ER Trabajo Economía 2018-IIДокумент2 страницы1ER Trabajo Economía 2018-IIhjbhhbjОценок пока нет
- Trabajo de CampoДокумент5 страницTrabajo de CampoLeydi Torcagua HuamaniОценок пока нет
- MonografiaДокумент78 страницMonografiaLeydi Torcagua HuamaniОценок пока нет
- Universidad Jose Carlos MariateguiДокумент9 страницUniversidad Jose Carlos MariateguiLeydi Torcagua HuamaniОценок пока нет
- EcologiasДокумент16 страницEcologiasLeydi Torcagua HuamaniОценок пока нет
- Universidad Jose Carlos MariateguiДокумент9 страницUniversidad Jose Carlos MariateguiLeydi Torcagua HuamaniОценок пока нет
- MonografiaДокумент78 страницMonografiaLeydi Torcagua HuamaniОценок пока нет
- MonografiaДокумент78 страницMonografiaLeydi Torcagua HuamaniОценок пока нет
- Presión HidrostáticaДокумент18 страницPresión HidrostáticaAnibalCahuaoОценок пока нет
- SINTESIS III PERIODO 6°. Leyes de NewtonДокумент8 страницSINTESIS III PERIODO 6°. Leyes de NewtonLUCIA OLIVEROSОценок пока нет
- Flujo A Traves de Fracturas y Canales IP-5511 Equipo 2Документ16 страницFlujo A Traves de Fracturas y Canales IP-5511 Equipo 2Hansel Ascencio SchmendrickОценок пока нет
- DESPIEZE GSH27 BOSCH Esquema + Listado PiezasДокумент7 страницDESPIEZE GSH27 BOSCH Esquema + Listado Piezasmanuel barberoОценок пока нет
- SifoneadoДокумент10 страницSifoneadoLuis JuarezОценок пока нет
- Factores de ConversiónДокумент2 страницыFactores de ConversiónDavid Stiven Ramirez MonsalveОценок пока нет
- Principios Básicos de ReologíaДокумент69 страницPrincipios Básicos de ReologíaElizaОценок пока нет
- Libro Diseno de Servicios Auxiliares CA-CC - CAEДокумент187 страницLibro Diseno de Servicios Auxiliares CA-CC - CAECarlos CastellonОценок пока нет
- Apuntes-Química A - 14 - Agosto-25 - Agosto-2023Документ15 страницApuntes-Química A - 14 - Agosto-25 - Agosto-2023Gaby AcostaОценок пока нет
- 396861-Article Text-577296-1-10-20220207Документ7 страниц396861-Article Text-577296-1-10-20220207Sergio VasquezОценок пока нет
- Ecuación de Energía MecánicaДокумент2 страницыEcuación de Energía MecánicaTania MirandaОценок пока нет
- Memoria Descriptiva Instalaciones SanitariasДокумент11 страницMemoria Descriptiva Instalaciones Sanitariascampoalegregranja6Оценок пока нет
- 2021 601 Bio Act 1 La Materia, Características GeneralesДокумент6 страниц2021 601 Bio Act 1 La Materia, Características GeneralesAndres Rayo Rayo CaballoОценок пока нет
- Unidad I Enero-2018Документ35 страницUnidad I Enero-2018Ivanny LucenaОценок пока нет
- Volumen Molar Fisicoquimica UmsaДокумент17 страницVolumen Molar Fisicoquimica UmsaGiselОценок пока нет
- PastorinoДокумент170 страницPastorinoRafael Armando ZaldañaОценок пока нет
- U1-S2 - Balance de EnergíaДокумент2 страницыU1-S2 - Balance de EnergíaJAVIER GONZALES VASQUEZОценок пока нет
- Práctica 3Документ2 страницыPráctica 3Isabella FerreiraОценок пока нет
- Deflexiones concreto armadoДокумент10 страницDeflexiones concreto armadoluispccomputerОценок пока нет
- Tarea Introduccion Del Estudio de La FisicaДокумент5 страницTarea Introduccion Del Estudio de La FisicaJose Tomas RodriguezОценок пока нет
- Fuerzas de Van Der WaalsДокумент4 страницыFuerzas de Van Der WaalsSailly Johana Saurith CandelarioОценок пока нет
- Fisica LuisaДокумент5 страницFisica LuisaFrancisco PaulinoОценок пока нет
- K-CC2-170-CONST-INF-025 - RB - EDW PoliclínicoДокумент40 страницK-CC2-170-CONST-INF-025 - RB - EDW PoliclínicoW.N. Ricci S.Оценок пока нет
- PronosticoДокумент3 страницыPronosticoABRAHAM NERY LLORENTY ROJASОценок пока нет
- Mapa Mental-Presion AtmosfericaДокумент3 страницыMapa Mental-Presion AtmosfericaFrancy MorenoОценок пока нет
- Informe Lab Quimica CalorimetriaДокумент13 страницInforme Lab Quimica CalorimetriaGisel0% (1)
- Diseño Camara de CargaДокумент5 страницDiseño Camara de CargaWalter Huatuco Lopez100% (1)