Вам также может понравиться
- Gene Editing, Epigenetic, Cloning and TherapyОт EverandGene Editing, Epigenetic, Cloning and TherapyРейтинг: 4 из 5 звезд4/5 (1)
- Epigenetics - Basic ConceptДокумент50 страницEpigenetics - Basic ConceptBiotech live92% (13)
- 2019 - Clinical EpigeneticsДокумент270 страниц2019 - Clinical EpigeneticsCharlsОценок пока нет
- Sun Woo Kang - Epigenetics, Environment, and Genes-Apple Academic Press (2013) PDFДокумент326 страницSun Woo Kang - Epigenetics, Environment, and Genes-Apple Academic Press (2013) PDFlaurenliddel100% (2)
- Epigenetics and NeuroendocrinologyДокумент280 страницEpigenetics and NeuroendocrinologyArianPedroza100% (3)
- 11b 1 The Living World010101Документ17 страниц11b 1 The Living World010101Deepak MojawatОценок пока нет
- Perceptions of EpigeneticsДокумент3 страницыPerceptions of Epigeneticsapi-20009652Оценок пока нет
- J00001 Oxford Int AS and A-Level Biology Specification WEB PDFДокумент46 страницJ00001 Oxford Int AS and A-Level Biology Specification WEB PDFPakistan English AcademyОценок пока нет
- Epigenetics in Health and DiseaseДокумент51 страницаEpigenetics in Health and DiseaseRashard Dyess-LaneОценок пока нет
- Wolpert, L. - 1994 - Do We Understand Development PDFДокумент2 страницыWolpert, L. - 1994 - Do We Understand Development PDFcontulmmivОценок пока нет
- Biochemistry Free For All FinalДокумент3 578 страницBiochemistry Free For All Finalharsh100% (1)
- Genetics TextbookДокумент2 553 страницыGenetics Textbookivanna salazar91% (11)
- Biology: Pearson Edexcel GCEДокумент8 страницBiology: Pearson Edexcel GCEyat yat szeОценок пока нет
- Essential Cell BiologyДокумент95 страницEssential Cell BiologyMahmoud Abdelrahman60% (5)
- Molecular Cell Biology 8th Edition Harvey Lodish2120 (WWW - Ebook DL - Com)Документ1 245 страницMolecular Cell Biology 8th Edition Harvey Lodish2120 (WWW - Ebook DL - Com)Sohl Santos96% (73)
- Optimal Foraging TheoryДокумент2 страницыOptimal Foraging TheoryJohn OsborneОценок пока нет
- As Level BiologyДокумент18 страницAs Level BiologyAasiya SultanaОценок пока нет
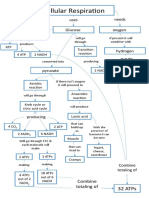
- Cellular Respiration Concept MapДокумент1 страницаCellular Respiration Concept MapmawakizakiОценок пока нет
- Pub - Microbiology and Biochemistry PDFДокумент271 страницаPub - Microbiology and Biochemistry PDFKuku328100% (1)
- Epigenetic Regulation of Gene Expression in CancerДокумент36 страницEpigenetic Regulation of Gene Expression in CancerSusi Rutmalem100% (2)
- 2013 Book EpigeneticsDevelopmentAndDisea PDFДокумент698 страниц2013 Book EpigeneticsDevelopmentAndDisea PDFSuelen Lima de Matos100% (1)
- Cell Structure and FunctionДокумент41 страницаCell Structure and FunctionAhkyluzLaniazОценок пока нет
- Science Single Cell BiologyДокумент204 страницыScience Single Cell BiologyAngel Slater100% (2)
- Genetics: How Are Traits Passed From Parents To Offspring?Документ40 страницGenetics: How Are Traits Passed From Parents To Offspring?ricardo4emeiaОценок пока нет
- Cell Biology and GeneticsДокумент144 страницыCell Biology and GeneticsReka Kutasi100% (1)
- From Embryology To Evo-Devo A History of Developmental Evolution (Dibner Institute Studies in The History of Science and Technology) by Manfred D. Laubichler (Editor), Jane Maienschein (Editor)Документ579 страницFrom Embryology To Evo-Devo A History of Developmental Evolution (Dibner Institute Studies in The History of Science and Technology) by Manfred D. Laubichler (Editor), Jane Maienschein (Editor)Mohammad Ali Sikder RamimОценок пока нет
- Neurohormonal Regulation of Body FunctionsДокумент34 страницыNeurohormonal Regulation of Body FunctionsjimmyОценок пока нет
- 28 Control of Blood Sugar Levels-SДокумент5 страниц28 Control of Blood Sugar Levels-SAaliya Nagori100% (1)
- 19 Evidence For Evolution-S PDFДокумент6 страниц19 Evidence For Evolution-S PDFstlcajun55Оценок пока нет
- Cell and Molecular Biology Module (Lecture and Laboratory)Документ200 страницCell and Molecular Biology Module (Lecture and Laboratory)RM Montemayor100% (2)
- Ib Bio Skills ApplicationsДокумент19 страницIb Bio Skills Applicationsapi-330898066Оценок пока нет
- Genetics PDFДокумент4 страницыGenetics PDFKhaled TurkОценок пока нет
- (Tertiary Level Biology) Peter Calow B.SC., Ph.D. (Auth.) - Evolutionary Principles-Springer US (1983)Документ116 страниц(Tertiary Level Biology) Peter Calow B.SC., Ph.D. (Auth.) - Evolutionary Principles-Springer US (1983)Trisan Kur100% (1)
- Costa (2010) - Horizons in Neuroscience ResearchДокумент452 страницыCosta (2010) - Horizons in Neuroscience Researchjonas1808Оценок пока нет
- BiologyДокумент31 страницаBiologyPrinceton University Press100% (1)
- Properties of Life: Biology Is The Science That Studies Life. What Exactly Is Life? This May Sound Like A SillyДокумент27 страницProperties of Life: Biology Is The Science That Studies Life. What Exactly Is Life? This May Sound Like A SillySandra Nicole RiveraОценок пока нет
- A2 Biology Exam QuestionsДокумент8 страницA2 Biology Exam QuestionseyhethОценок пока нет
- Introduction To Biomolecular Structure and Biophysics Basics of BiophysicsДокумент282 страницыIntroduction To Biomolecular Structure and Biophysics Basics of BiophysicsAli Mariouni Alawie100% (1)
- Molecular Biology of The Cell, 5th EditionДокумент82 страницыMolecular Biology of The Cell, 5th EditionBee Nunes24% (66)
- Answering Exam Questions On CellsДокумент4 страницыAnswering Exam Questions On CellsTamicka Bonnick100% (1)
- LS2 Study GuideДокумент79 страницLS2 Study GuideJaneОценок пока нет
- Cell Biology Pollard 2e PDFДокумент927 страницCell Biology Pollard 2e PDFМарина Попозогло83% (6)
- Neuroscience Unravelling The Mysteries of The Brain: For Students A CareerДокумент8 страницNeuroscience Unravelling The Mysteries of The Brain: For Students A CareerNancy Angélica LopezОценок пока нет
- Commonlit Teenage Brains Are Malleable and Vulnerable Researchers SayДокумент5 страницCommonlit Teenage Brains Are Malleable and Vulnerable Researchers Sayapi-506044294Оценок пока нет
- Flashcards - CP 18 Production of Amylase in Germinating Cereal Grains - Edexcel Biology International A-LevelДокумент15 страницFlashcards - CP 18 Production of Amylase in Germinating Cereal Grains - Edexcel Biology International A-LevelBara' Hammadeh0% (1)
- CH 08Документ81 страницаCH 08Divine Grace InovejasОценок пока нет
- 2014 Handbook of NeurotoxicityДокумент2 360 страниц2014 Handbook of NeurotoxicityAlberth Kamus100% (1)
- Biology Mapping GuideДокумент28 страницBiology Mapping GuideGazar100% (1)
- Epigenetic SДокумент8 страницEpigenetic Smaria dulce100% (1)
- Nearpeer MDCAT Biology 2018Документ733 страницыNearpeer MDCAT Biology 2018ayesha amjadОценок пока нет
- Hot Topics in Cell BiologyДокумент279 страницHot Topics in Cell BiologyChartridge Books Oxford100% (2)
- 083 - Chromatography and Its Uses in BiologyДокумент3 страницы083 - Chromatography and Its Uses in Biologylastjoe71Оценок пока нет
- An Introduction To EpigeneticsДокумент8 страницAn Introduction To EpigeneticsSyncOrSwim83% (6)
- (Brown, Gene Cloning and DNA Analysis) Terry Brown - Gene Cloning and DNA Analysis - An Introduction (Brown, Gene Cloning and DNA Analysis) - Wiley-Blackwell (2010)Документ338 страниц(Brown, Gene Cloning and DNA Analysis) Terry Brown - Gene Cloning and DNA Analysis - An Introduction (Brown, Gene Cloning and DNA Analysis) - Wiley-Blackwell (2010)nefko 42083% (6)
- EE - IB Extended Essay Topic Ideas FOR BIOLOGYДокумент4 страницыEE - IB Extended Essay Topic Ideas FOR BIOLOGYArshiya TanveerОценок пока нет
- BiologyДокумент984 страницыBiologyGary Fearless Edwards86% (7)
- Reproduction and Sexuality in Marine Fishes: Patterns and ProcessesОт EverandReproduction and Sexuality in Marine Fishes: Patterns and ProcessesKathleen S. ColeОценок пока нет
- Cell Growth and Cell DivisionОт EverandCell Growth and Cell DivisionR. J. C. HarrisРейтинг: 5 из 5 звезд5/5 (1)
- Edexcel IAL A2 Chemistry New Textbook Sample PagesДокумент41 страницаEdexcel IAL A2 Chemistry New Textbook Sample PagesGazarОценок пока нет
- Ial Chemistry (2018) Getting-Started-GuideДокумент25 страницIal Chemistry (2018) Getting-Started-GuideGazar0% (1)
- L27 VasculitisДокумент28 страницL27 VasculitisGazarОценок пока нет
- Edexcel IAL Decision Maths 1 Sample PagesДокумент102 страницыEdexcel IAL Decision Maths 1 Sample PagesGazar100% (3)
- Biology Mapping GuideДокумент28 страницBiology Mapping GuideGazar100% (1)
- Chemistry Teacher Mathematics Support GuideДокумент34 страницыChemistry Teacher Mathematics Support GuideGazar100% (1)
- Biology Student Mathematics Support GuideДокумент42 страницыBiology Student Mathematics Support GuideGazarОценок пока нет
- Edexcel IAL Biology Lab Book PDFДокумент52 страницыEdexcel IAL Biology Lab Book PDFGazar67% (18)
- Chemistry Student Practical GuideДокумент26 страницChemistry Student Practical GuideGazar50% (2)
- Chemistry Teacher Practical GuideДокумент42 страницыChemistry Teacher Practical GuideGazar100% (5)
- Chemistry Topic Guide Instrumental AnalysisДокумент30 страницChemistry Topic Guide Instrumental AnalysisGazarОценок пока нет
- Edexcel IAL Chemistry Lab BookДокумент26 страницEdexcel IAL Chemistry Lab BookGazar79% (19)
- As Exemplars With CommentariesДокумент24 страницыAs Exemplars With CommentariesGazarОценок пока нет
- Chemistry Transition Guide Containing Worksheets, Examiner Reports, Exam Practice EtcДокумент90 страницChemistry Transition Guide Containing Worksheets, Examiner Reports, Exam Practice EtcGazar100% (1)
- Chemistry Topic Guide Energetics Energy and EntropyДокумент21 страницаChemistry Topic Guide Energetics Energy and EntropyGazar100% (1)
- Biology Practical Guide TeachersДокумент37 страницBiology Practical Guide TeachersGazar88% (8)
- IAS Chemistry Student Book 1 (2018) AnswersДокумент53 страницыIAS Chemistry Student Book 1 (2018) AnswersGazar61% (119)
- Edexcel IAL Physics Lab BookДокумент30 страницEdexcel IAL Physics Lab BookGazar61% (18)
- IAS Biology Student Book 1 (2018) AnswersДокумент74 страницыIAS Biology Student Book 1 (2018) AnswersGazar76% (33)
- IAS Physics Student Book 1 (2018) AnswersДокумент32 страницыIAS Physics Student Book 1 (2018) AnswersGazar77% (56)
- Test Bank For Molecular Biology Principles and Practice 1st Edition Michael M CoxДокумент9 страницTest Bank For Molecular Biology Principles and Practice 1st Edition Michael M CoxAnthonyRogersydtfp100% (64)
- Harper's Illustrated Biochemistry CH 35Документ11 страницHarper's Illustrated Biochemistry CH 35Richelle Dianne Ramos-Giang100% (1)
- Animal Biology Biotechnology 2019Документ13 страницAnimal Biology Biotechnology 2019Vaishnavi ReddyОценок пока нет
- C 06 From Chromosomes To GenomesДокумент60 страницC 06 From Chromosomes To GenomesHamadaShehataОценок пока нет
- Question Bank - Topic 7 Nucleic AcidsДокумент49 страницQuestion Bank - Topic 7 Nucleic AcidsMuliasena NormadianОценок пока нет
- Chương 1 - GenomicДокумент32 страницыChương 1 - GenomicNguyễn Hữu Bảo MinhОценок пока нет
- Genomics and ProteomicsДокумент317 страницGenomics and ProteomicsIma AnОценок пока нет
- Review Questions For Human HistologyДокумент438 страницReview Questions For Human Histologyjoeguy6986% (7)
- Chromatin Remodeling Complexes - The Regulators of Genome FunctionДокумент19 страницChromatin Remodeling Complexes - The Regulators of Genome FunctionManisha DasОценок пока нет
- Thompson Thompson Genetics in Medicine e Book Thompson and Thompson Genetics in Medicine 7th Edition Ebook PDFДокумент62 страницыThompson Thompson Genetics in Medicine e Book Thompson and Thompson Genetics in Medicine 7th Edition Ebook PDFjack.murgia175100% (32)
- Genes and Chromosomes: Lehninger. Principles of Biochemistry. by Nelson and Cox, 5 Edition W.H. Freeman and CompanyДокумент121 страницаGenes and Chromosomes: Lehninger. Principles of Biochemistry. by Nelson and Cox, 5 Edition W.H. Freeman and CompanyAmeylia KrisОценок пока нет
- Lehninger PPT Ch24Документ61 страницаLehninger PPT Ch24Nia IarajuliОценок пока нет
- Snep PDFДокумент13 страницSnep PDFNaga RajanОценок пока нет
- 10.4: Eukaryotic Gene RegulationДокумент7 страниц10.4: Eukaryotic Gene RegulationFlorence Gaone GaongalelweОценок пока нет
- 2013 Book EpigeneticsDevelopmentAndDisea PDFДокумент698 страниц2013 Book EpigeneticsDevelopmentAndDisea PDFSuelen Lima de Matos100% (1)
- DNA Structure and Information TransferДокумент19 страницDNA Structure and Information TransferKarmina SantosОценок пока нет
- 2 Genetics Lecture - DNA PackagingДокумент45 страниц2 Genetics Lecture - DNA PackagingAMIRA HELAYELОценок пока нет
- Structure and Organization of ChromatinДокумент11 страницStructure and Organization of Chromatincassidy conchaОценок пока нет
- Chromosomes, DNA Structure and TopologyДокумент92 страницыChromosomes, DNA Structure and TopologyShalini Muthu100% (2)
- Nature Publishing Group - Epigenetics CollectionДокумент70 страницNature Publishing Group - Epigenetics CollectionSounak Ghosh Roy100% (1)
- Aiims - Biochemistry Q&AДокумент198 страницAiims - Biochemistry Q&APrahladRao KulkarniОценок пока нет
- Ch10-1 Gen MaterialДокумент36 страницCh10-1 Gen MaterialarcheologistsОценок пока нет
- How Does Epigenetics Influence The Course of Evolution?: Alyson Ashe, Vincent Colot and Benjamin P. OldroydДокумент9 страницHow Does Epigenetics Influence The Course of Evolution?: Alyson Ashe, Vincent Colot and Benjamin P. OldroydAndrea Caicedo DiazОценок пока нет
- Epigenetics in Cancer: Review ArticleДокумент12 страницEpigenetics in Cancer: Review ArticleItzel MercadoОценок пока нет
- Structure of Eukaryotic ChromosomeДокумент29 страницStructure of Eukaryotic ChromosomerohishaakОценок пока нет
- Organization Replication Repair: DR S K BansalДокумент71 страницаOrganization Replication Repair: DR S K BansalSanjiv BansalОценок пока нет
- CytogeneticsДокумент9 страницCytogeneticsnaiktariq00Оценок пока нет
- Prokaryotic Genome OrganizationДокумент53 страницыProkaryotic Genome OrganizationAqsa YaminОценок пока нет
- Cell and Human Chromosomes 17Документ46 страницCell and Human Chromosomes 17poojaОценок пока нет
- Genome Organization and ControlДокумент32 страницыGenome Organization and Controllmiguel92100% (1)