Вам также может понравиться
- Substâncias húmicas aquáticas: Interações com espécies metálicasОт EverandSubstâncias húmicas aquáticas: Interações com espécies metálicasОценок пока нет
- (Apostila) Cromatografia - Ufpe PDFДокумент81 страница(Apostila) Cromatografia - Ufpe PDFusinavaleОценок пока нет
- Alexandre Schuler - CromatografiaДокумент70 страницAlexandre Schuler - CromatografiaArleteОценок пока нет
- Cromatografia - A Gás e A Líquido (SCHULER, A.) PDFДокумент82 страницыCromatografia - A Gás e A Líquido (SCHULER, A.) PDFLuan Gabriel100% (1)
- Cromatografia Contracorrente PDFДокумент107 страницCromatografia Contracorrente PDFJulia XimenesОценок пока нет
- Cromatografia em ContracorrenteДокумент37 страницCromatografia em ContracorrenteBruno Giordano100% (1)
- Cromatografia Contra-Corrente PDFДокумент30 страницCromatografia Contra-Corrente PDFAntony DpbОценок пока нет
- Lista de Exercícios-EuzébioДокумент17 страницLista de Exercícios-EuzébioIsisTОценок пока нет
- Apostila Aulas Praticas de Quimica Inorganica IIIДокумент22 страницыApostila Aulas Praticas de Quimica Inorganica IIIPatryck SassakiОценок пока нет
- Catálise QuímicaДокумент33 страницыCatálise QuímicaLeonardo MacielОценок пока нет
- Impacto Izod ASTM D256Документ4 страницыImpacto Izod ASTM D256Luis Sidnei MachadoОценок пока нет
- Síntese Do Sulfato de Tetraminocobre (II) Mono-HidratadoДокумент6 страницSíntese Do Sulfato de Tetraminocobre (II) Mono-HidratadoPatrícia AzinheiraОценок пока нет
- Aula 5 - ExercíciosДокумент9 страницAula 5 - ExercíciosWil BarbosaОценок пока нет
- Relatório 8 Cromatografia CCDДокумент6 страницRelatório 8 Cromatografia CCDGuilherme MarquesОценок пока нет
- Apostila de CromatografiaДокумент23 страницыApostila de CromatografiaDanielle Barbosa100% (1)
- Espectrômetros e EspectrofotômetrosДокумент30 страницEspectrômetros e EspectrofotômetrosMichell SzczpanikОценок пока нет
- Relatorio - Ácidos e BasesДокумент6 страницRelatorio - Ácidos e BasesJoão Pedro VasconcelosОценок пока нет
- Cromatografia Gasosa Apostila GeralДокумент122 страницыCromatografia Gasosa Apostila Geralredcat1233Оценок пока нет
- 05 - Espectrometria de Absorção Atomica 2Документ58 страниц05 - Espectrometria de Absorção Atomica 2Cecília CarvalhoОценок пока нет
- Laboratório de Química OrgânicaДокумент30 страницLaboratório de Química OrgânicaKoca OliveiraОценок пока нет
- CromatografiaДокумент19 страницCromatografiaJ. MessiasОценок пока нет
- PTG 2019Документ26 страницPTG 2019lacelrjОценок пока нет
- Determinação de Constantes Físicas de Compostos OrgânicosДокумент12 страницDeterminação de Constantes Físicas de Compostos OrgânicosSabrina Mara100% (1)
- Coluna Destila - o Pratos PerfuradosДокумент11 страницColuna Destila - o Pratos PerfuradosIaraFerreiradeRezendeОценок пока нет
- Aplicação Dos Vetores No Estudo Da Polaridade de MoléculasДокумент4 страницыAplicação Dos Vetores No Estudo Da Polaridade de MoléculasRogerio AndradeОценок пока нет
- Relatorio 04 CalorДокумент10 страницRelatorio 04 Caloremanuel linoОценок пока нет
- PCR em Tempo RealДокумент48 страницPCR em Tempo RealJorge SalinasОценок пока нет
- Cromatografia de Camada DelgadaДокумент15 страницCromatografia de Camada DelgadaLaisaPeraciniОценок пока нет
- Experimento de Reações Químicas e Troca de EnergiaДокумент3 страницыExperimento de Reações Químicas e Troca de EnergiaIndividualista DuelistaОценок пока нет
- Roteiro Do Experimento Balança de Pratos - EstáticaДокумент9 страницRoteiro Do Experimento Balança de Pratos - EstáticaadmcavaliniassessoriaОценок пока нет
- Cromatografia Liquida - Metodos CromatograficosДокумент93 страницыCromatografia Liquida - Metodos CromatograficosbncamelloОценок пока нет
- Cromatografia em PapelДокумент6 страницCromatografia em PapelJoão Henrique Ozon100% (1)
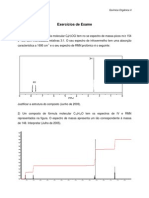
- Exercicios de ExameДокумент3 страницыExercicios de ExameAlberto GomesОценок пока нет
- Análise Quimica Dos Compostos Bioativos Da Pvermelha 2017 PDFДокумент120 страницAnálise Quimica Dos Compostos Bioativos Da Pvermelha 2017 PDFJosenilton BomfimОценок пока нет
- Acido AscorbicoДокумент6 страницAcido AscorbicoMarleneFernandes100% (1)
- Exercicios 2-Tratamento de ÁguaДокумент2 страницыExercicios 2-Tratamento de ÁguaLarissa0610 braz100% (1)
- Relatório de Física - Geradores ElétricosДокумент10 страницRelatório de Física - Geradores ElétricosEduarda de Matos Figueiredo100% (1)
- Lista Exercícios FenômenosДокумент17 страницLista Exercícios Fenômenosmarcelodalbo0% (1)
- Titulac - A - o Potenciometrica Da GlicinaДокумент15 страницTitulac - A - o Potenciometrica Da GlicinaCarla Nogueira0% (1)
- Exercicios Resolvidos Capitulo 8 SolomonsДокумент11 страницExercicios Resolvidos Capitulo 8 SolomonsdefatimapedroОценок пока нет
- Relatório Estágio 2 PDFДокумент21 страницаRelatório Estágio 2 PDFCarmen AndrettaОценок пока нет
- Ponto de Fusão de Compostos Orgânicos (Alfa Naftol e Acido Benzóico)Документ13 страницPonto de Fusão de Compostos Orgânicos (Alfa Naftol e Acido Benzóico)Ricardo LimaОценок пока нет
- Lista de Exercícios 3Документ2 страницыLista de Exercícios 3MatheusLopes100% (1)
- Experimento 4 Equilbrio Quimico RTE2021Документ5 страницExperimento 4 Equilbrio Quimico RTE2021Leticia BrazОценок пока нет
- 061lista2 JoaoFelipe GabaritoДокумент24 страницы061lista2 JoaoFelipe GabaritoBruno GiglioОценок пока нет
- Fundamentos HPLC Curso 2014 Teoria e PráticaДокумент146 страницFundamentos HPLC Curso 2014 Teoria e PráticaJoão Wilson Filho100% (1)
- Voltametria AssuntoДокумент58 страницVoltametria AssuntoLarissa AraújoОценок пока нет
- Relatório #3 - Teor de AmôniaДокумент3 страницыRelatório #3 - Teor de AmôniaKurtz100% (1)
- Sintese Da BenzocaínaДокумент16 страницSintese Da BenzocaínaLeandronmsmОценок пока нет
- AbsorciometriaДокумент8 страницAbsorciometriaLarissa LorennОценок пока нет
- Aula 11 Cromatografia Líquida 2S2012Документ24 страницыAula 11 Cromatografia Líquida 2S2012Leonardo MacielОценок пока нет
- 1 - Processos Industriais Organicos PDFДокумент15 страниц1 - Processos Industriais Organicos PDFDiego TeixeiraОценок пока нет
- Relatório I - Curva de AbsorçãoДокумент11 страницRelatório I - Curva de AbsorçãodavitersouzaОценок пока нет
- Química Inorgânica Descritiva - Bloco P - 15a17Документ87 страницQuímica Inorgânica Descritiva - Bloco P - 15a17BrunoRamosdeLimaОценок пока нет
- Cromatografia (Livro)Документ68 страницCromatografia (Livro)SIMAO ALFREDO THEMBANEОценок пока нет
- Cromatografia (Livro)Документ66 страницCromatografia (Livro)Kelen Cristina Vivan Bilo100% (3)
- Relatório - AdsorçãoДокумент21 страницаRelatório - AdsorçãoaccfjОценок пока нет
- Relatório Cromatografia Gasosa Completo - Grupofodaorestoémoda - Qui8mДокумент12 страницRelatório Cromatografia Gasosa Completo - Grupofodaorestoémoda - Qui8mBruno FariasОценок пока нет
- Relatório 3 UFPE Química Geral ExperimentalДокумент5 страницRelatório 3 UFPE Química Geral ExperimentalCésar CanejaОценок пока нет
- Relatorio CromatografiaДокумент7 страницRelatorio CromatografiaBeatriz LimaОценок пока нет
- Palestra Compostos Bioativos Da Soja - SBAF Junho 2008Документ52 страницыPalestra Compostos Bioativos Da Soja - SBAF Junho 2008Denise Esteves MoritzОценок пока нет
- Análise Sensorial Dos AlimentosДокумент25 страницAnálise Sensorial Dos AlimentosDenise Esteves MoritzОценок пока нет
- Alexandre Schuler CADERNO DE LABORATÓRIO DE CROMATOGRAFIAДокумент15 страницAlexandre Schuler CADERNO DE LABORATÓRIO DE CROMATOGRAFIADenise Esteves MoritzОценок пока нет
- 02 Ferm Alcoolica Montagem FermentadorДокумент2 страницы02 Ferm Alcoolica Montagem FermentadorDenise Esteves MoritzОценок пока нет
- Embrapa Controle Qualidade Doc-158-LeiteДокумент24 страницыEmbrapa Controle Qualidade Doc-158-LeiteDenise Esteves MoritzОценок пока нет
- Alterações Alimentares Tec AlimentosДокумент38 страницAlterações Alimentares Tec AlimentosDenise Esteves Moritz100% (1)
- Biotecnologia 2016 PDFДокумент312 страницBiotecnologia 2016 PDFKeli Sobral100% (1)
- Relatório 10 - Carvão AtivadoДокумент4 страницыRelatório 10 - Carvão AtivadoRailaneMonteiroОценок пока нет
- Relatório de AdsorçãoДокумент23 страницыRelatório de AdsorçãoThamyres CôcoОценок пока нет
- Físico-Química - CinéticaДокумент3 страницыFísico-Química - CinéticaPaulo GonçalvesОценок пока нет
- 2 Lista Mecânica Estatística - 2o. 2023Документ4 страницы2 Lista Mecânica Estatística - 2o. 2023joao.astolfoОценок пока нет
- Roberto Heeman - Carbonífera CatarinenseДокумент41 страницаRoberto Heeman - Carbonífera CatarinenseARTUR VICARI GRANATOОценок пока нет
- AmiloglicosilaseДокумент7 страницAmiloglicosilaseRaíssa RibeiroОценок пока нет
- 10-Geotecania AmbientalДокумент7 страниц10-Geotecania AmbientalBruna SilveiraОценок пока нет
- Determinação Da Curva de Defloculação PDFДокумент16 страницDeterminação Da Curva de Defloculação PDFJ. GirotoОценок пока нет
- Análise Comparativa Da Secagem Do Bagaço de Cana Utilizando Secador Elétrico e Secador SolarДокумент106 страницAnálise Comparativa Da Secagem Do Bagaço de Cana Utilizando Secador Elétrico e Secador SolarRômulo SanzioОценок пока нет
- Impermeabilização de Pisos Enterrados: March 2000Документ90 страницImpermeabilização de Pisos Enterrados: March 2000heldersilvaferreira2264Оценок пока нет
- SHP - Aula - 2 - Cont PneumaticaДокумент61 страницаSHP - Aula - 2 - Cont PneumaticaJoão Vitor Santos SilvaОценок пока нет
- Lista 2 - ResolvidaДокумент10 страницLista 2 - ResolvidaJunin Oliveira100% (2)
- Dessulfurização de BiogasДокумент27 страницDessulfurização de BiogasDixon AvelinoОценок пока нет
- 0 Relatório AdsorçãoДокумент15 страниц0 Relatório AdsorçãoJill FordОценок пока нет
- Descoloração de E Uente Têxtil Por AdsorçãoДокумент34 страницыDescoloração de E Uente Têxtil Por AdsorçãoEduarda BungnerОценок пока нет
- 03 Cromatografia Coluna ClassicaДокумент5 страниц03 Cromatografia Coluna ClassicaPatrícia Felix ÁvilaОценок пока нет
- Noções Gerais de Anodização PDFДокумент54 страницыNoções Gerais de Anodização PDFluciaОценок пока нет
- 328Документ28 страниц328Alessandro BenevidesОценок пока нет
- Argilas Bentoníticas: Conceitos, Estruturas, Propriedades, Usos Industriais, Reservas, Produção e Produtores/fornecedores Nacionais e InternacionaisДокумент10 страницArgilas Bentoníticas: Conceitos, Estruturas, Propriedades, Usos Industriais, Reservas, Produção e Produtores/fornecedores Nacionais e InternacionaisDimi RandelОценок пока нет
- Técnica de BETДокумент31 страницаTécnica de BETNelson_Lage100% (1)
- Tratamento Terciário de Efluentes de Refinaria de Petróleo Com Carvão Ativado em PóДокумент76 страницTratamento Terciário de Efluentes de Refinaria de Petróleo Com Carvão Ativado em PóZ3tyyTVОценок пока нет
- Lista Dos Exercicios.2docxДокумент3 страницыLista Dos Exercicios.2docxDian AlbertoОценок пока нет
- Gases e Vapores PDFДокумент16 страницGases e Vapores PDFJuliano André PetryОценок пока нет
- Físico-Química II Aula Fenômenos de Superfície IДокумент21 страницаFísico-Química II Aula Fenômenos de Superfície ILuddy MendesОценок пока нет
- Quimica Hibridacao FIM PeriodicidadeДокумент82 страницыQuimica Hibridacao FIM PeriodicidadePEDRO FERNANDESОценок пока нет
- Noções de Operações UnitáriasДокумент3 страницыNoções de Operações UnitáriasAdolpho Ramsés0% (1)
- Análise Da Escoabilidade de PósДокумент129 страницAnálise Da Escoabilidade de PósmapelliserОценок пока нет