Вам также может понравиться
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- CVLattes English M01 D18Документ6 страницCVLattes English M01 D18jose mirandaОценок пока нет
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- TanbourinemanДокумент3 страницыTanbourinemanjose mirandaОценок пока нет
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- Flow Node RedДокумент10 страницFlow Node Redjose mirandaОценок пока нет
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- Coverletterin EnglishДокумент1 страницаCoverletterin Englishjose mirandaОценок пока нет
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- Cifra Club - George Harrison - My Sweet Lord PDFДокумент6 страницCifra Club - George Harrison - My Sweet Lord PDFjose mirandaОценок пока нет
- CVLattes English M01 D18Документ6 страницCVLattes English M01 D18jose mirandaОценок пока нет
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- General Physics 1: The Commission On Higher EducationДокумент130 страницGeneral Physics 1: The Commission On Higher EducationRaidis Dela CruzОценок пока нет
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- Tabel A Deder I VadasДокумент1 страницаTabel A Deder I Vadasjose mirandaОценок пока нет
- Wave Scattered Lau e BraggДокумент17 страницWave Scattered Lau e Braggjose mirandaОценок пока нет
- Bead On Rotating HoopДокумент3 страницыBead On Rotating Hoopjose mirandaОценок пока нет
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- SchrodingerДокумент56 страницSchrodingerIka RisnawatiОценок пока нет
- MatlabcodesДокумент26 страницMatlabcodesjose mirandaОценок пока нет
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- George Harrison - My Sweet LordДокумент6 страницGeorge Harrison - My Sweet Lordjose mirandaОценок пока нет
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- CalculosfisicaДокумент2 страницыCalculosfisicajose mirandaОценок пока нет
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (344)
- CalculosfisicДокумент4 страницыCalculosfisicjose mirandaОценок пока нет
- Electromagnetic Wave Lesson PlanДокумент2 страницыElectromagnetic Wave Lesson Planjose miranda100% (1)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- Collaborative WritingДокумент2 страницыCollaborative Writingjose miranda100% (1)
- Crystal StructureДокумент14 страницCrystal StructureMahesh Lohith K.S100% (4)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- Ode Lesson PlanДокумент9 страницOde Lesson Planjose miranda0% (1)
- Dielectric Properties of SolidsДокумент15 страницDielectric Properties of SolidsMahesh Lohith K.S100% (11)
- Escola Primária Teacher: (XXX) XXX-XXXX 142 Your Address BLVD, City Name, CA XXXXXДокумент2 страницыEscola Primária Teacher: (XXX) XXX-XXXX 142 Your Address BLVD, City Name, CA XXXXXjose mirandaОценок пока нет
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- Einstein (2018)Документ500 страницEinstein (2018)jose mirandaОценок пока нет
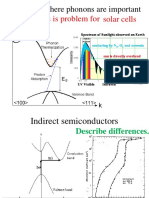
- Solar Cells Energy Loss Is Problem For: Examples Where Phonons Are ImportantДокумент51 страницаSolar Cells Energy Loss Is Problem For: Examples Where Phonons Are Importantjose mirandaОценок пока нет
- Lesson Plan #28: Functions? 1) Students Will Be Able To Differentiate Trigonometric FunctionsДокумент5 страницLesson Plan #28: Functions? 1) Students Will Be Able To Differentiate Trigonometric Functionsjose mirandaОценок пока нет
- Presentation DielectricДокумент25 страницPresentation Dielectricjose mirandaОценок пока нет
- Post Doc ProposalДокумент5 страницPost Doc Proposaljose mirandaОценок пока нет
- Superconductivity: Peter SCHM UserДокумент47 страницSuperconductivity: Peter SCHM Userjose mirandaОценок пока нет
- Optical Electrical Blood Vital SensorsДокумент37 страницOptical Electrical Blood Vital Sensorsjose mirandaОценок пока нет
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- Lesson Plan: MagnetismДокумент25 страницLesson Plan: Magnetismjose mirandaОценок пока нет
- Specific Objectives:: Unit 5: Limit and DerivativeДокумент1 страницаSpecific Objectives:: Unit 5: Limit and Derivativejose mirandaОценок пока нет
- Second Term English Exam: Level TCST June 2021Документ6 страницSecond Term English Exam: Level TCST June 2021benfaresОценок пока нет
- G 62 - 14 PDFДокумент4 страницыG 62 - 14 PDFjose floresОценок пока нет
- Architecture of HimalayasДокумент3 страницыArchitecture of HimalayasAndrea CaballeroОценок пока нет
- An Automated Energy Meter Reading System Using GSM TechnologyДокумент8 страницAn Automated Energy Meter Reading System Using GSM TechnologyBarОценок пока нет
- Hemodynamic Monitoring in ICUДокумент111 страницHemodynamic Monitoring in ICUManjunath Gemini100% (2)
- Full Site PDFДокумент23 страницыFull Site PDFpursuwОценок пока нет
- DinmjgДокумент10 страницDinmjghaker linkisОценок пока нет
- Sample Paper English: Kendriya Vidyalaya SangathanДокумент7 страницSample Paper English: Kendriya Vidyalaya SangathanVines and ScienceОценок пока нет
- 18.1 Outline The Mechanisms Which: Chemotherapy Target Dividing CellsДокумент8 страниц18.1 Outline The Mechanisms Which: Chemotherapy Target Dividing CellsSenthereng MoaisiОценок пока нет
- EPTT5100 - Pressure - Temperature Sensor - 1308 - GДокумент2 страницыEPTT5100 - Pressure - Temperature Sensor - 1308 - GHendry Putra RahadiОценок пока нет
- Kelvin Hughes LTD: Technical Advice SheetДокумент7 страницKelvin Hughes LTD: Technical Advice SheetVladymirОценок пока нет
- Unit 21Документ22 страницыUnit 21Yuni IndahОценок пока нет
- Uptime KitsДокумент3 страницыUptime KitsMtto Materia PrimaОценок пока нет
- TP260SR Tier 3 TC002-1037Документ1 страницаTP260SR Tier 3 TC002-1037Jorge GalarceОценок пока нет
- Dave Graham Literature CatalogДокумент640 страницDave Graham Literature CatalogPierce PetersonОценок пока нет
- RB in Poultry Feed - 3Документ17 страницRB in Poultry Feed - 3Vishwanath HebbiОценок пока нет
- DP16B Bench Drill PressДокумент20 страницDP16B Bench Drill Pressalfri7370% (1)
- Type of TrucksДокумент8 страницType of TrucksYojhan VelezОценок пока нет
- Content (SG)Документ88 страницContent (SG)Kusuma Cakra WardayaОценок пока нет
- Easy Guide For Fujitsu T901 LaptopДокумент141 страницаEasy Guide For Fujitsu T901 LaptopElaineОценок пока нет
- CP Inf4Документ357 страницCP Inf4Rugwed JadhavОценок пока нет
- AngelДокумент21 страницаAngelNoj ZachОценок пока нет
- BKC 80Документ2 страницыBKC 80jawaidchemicalsОценок пока нет
- IJHIM 6 - Nur Husnina (36 SD 42)Документ7 страницIJHIM 6 - Nur Husnina (36 SD 42)RSU Sayang BundaОценок пока нет
- B11 - Overload Relays (Ref) ENДокумент20 страницB11 - Overload Relays (Ref) ENAhmed AbazaОценок пока нет
- Yu-Gi-Oh GX Duel Academy - Written ExamДокумент26 страницYu-Gi-Oh GX Duel Academy - Written ExamisishamalielОценок пока нет
- REM Geo&Man Comp 2016Документ350 страницREM Geo&Man Comp 2016youungОценок пока нет
- Part PabrikДокумент2 страницыPart PabrikNaldy NaldyОценок пока нет
- Parasites in Reptile PDFДокумент21 страницаParasites in Reptile PDFRamadhani SyafitriОценок пока нет
- Sediments and Sedimentary Rock-Week 4Документ61 страницаSediments and Sedimentary Rock-Week 4qomaruzzaman5740Оценок пока нет