Вам также может понравиться
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryОт EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryРейтинг: 3.5 из 5 звезд3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)От EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Рейтинг: 4.5 из 5 звезд4.5/5 (121)
- Never Split the Difference: Negotiating As If Your Life Depended On ItОт EverandNever Split the Difference: Negotiating As If Your Life Depended On ItРейтинг: 4.5 из 5 звезд4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingОт EverandThe Little Book of Hygge: Danish Secrets to Happy LivingРейтинг: 3.5 из 5 звезд3.5/5 (400)
- Kawasaki Bayou 300 Service Manual RepairДокумент678 страницKawasaki Bayou 300 Service Manual RepairMac Thon81% (64)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaОт EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaРейтинг: 4.5 из 5 звезд4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeОт EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeРейтинг: 4 из 5 звезд4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreОт EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreРейтинг: 4 из 5 звезд4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyОт EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyРейтинг: 3.5 из 5 звезд3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersОт EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersРейтинг: 4.5 из 5 звезд4.5/5 (345)
- The Emperor of All Maladies: A Biography of CancerОт EverandThe Emperor of All Maladies: A Biography of CancerРейтинг: 4.5 из 5 звезд4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnОт EverandTeam of Rivals: The Political Genius of Abraham LincolnРейтинг: 4.5 из 5 звезд4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceОт EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceРейтинг: 4 из 5 звезд4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureОт EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureРейтинг: 4.5 из 5 звезд4.5/5 (474)
- The Yellow House: A Memoir (2019 National Book Award Winner)От EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Рейтинг: 4 из 5 звезд4/5 (98)
- The Unwinding: An Inner History of the New AmericaОт EverandThe Unwinding: An Inner History of the New AmericaРейтинг: 4 из 5 звезд4/5 (45)
- BOP Choke Man Inspection ProcedureДокумент13 страницBOP Choke Man Inspection ProcedureAhmed Imtiaz Rao100% (2)
- Caterpillar Taurus 70 Users Manual 536177 PDFДокумент83 страницыCaterpillar Taurus 70 Users Manual 536177 PDFTrung Quan Vo100% (1)
- Nonlinear Buckling of Micropiles FinalДокумент15 страницNonlinear Buckling of Micropiles FinalcenglertОценок пока нет
- PIPE STRESS ANALYSIS - FIV - AIV (Compatibility Mode)Документ48 страницPIPE STRESS ANALYSIS - FIV - AIV (Compatibility Mode)salma khalilaa putrii 6D100% (1)
- TECHNICAL NOTES (GENERAL SPECIFICATION) FOR VALVES (Gate, Globe, Check, Ball, Plug, Needle, Butterfly & Piston Valves)Документ29 страницTECHNICAL NOTES (GENERAL SPECIFICATION) FOR VALVES (Gate, Globe, Check, Ball, Plug, Needle, Butterfly & Piston Valves)Nilesh Mistry100% (2)
- COMPABLOC CoolerДокумент37 страницCOMPABLOC CoolerAntonОценок пока нет
- Piping Design and Detailed EngineeringДокумент3 страницыPiping Design and Detailed EngineeringsdОценок пока нет
- Experimental Study of Batch Reactor Performance For Ethyl Acetate SaponificationДокумент13 страницExperimental Study of Batch Reactor Performance For Ethyl Acetate SaponificationDwinaRahmayaniОценок пока нет
- Piping Course 1Документ97 страницPiping Course 1AfetОценок пока нет
- (IOP Concise Physics) Liengme, Bernard V - Excel VBA For Physicists A Primer-IOP Publishing (2016)Документ92 страницы(IOP Concise Physics) Liengme, Bernard V - Excel VBA For Physicists A Primer-IOP Publishing (2016)DwinaRahmayani100% (1)
- Alignment Coupling Operation Manual Tyre-FlexДокумент37 страницAlignment Coupling Operation Manual Tyre-Flexer_sanjaypatelОценок пока нет
- Cryogenic TurboexpandersДокумент9 страницCryogenic TurboexpandersDwinaRahmayaniОценок пока нет
- Orange Juice: Guideline ForДокумент13 страницOrange Juice: Guideline ForDwinaRahmayaniОценок пока нет
- International Journal of TechnologyДокумент12 страницInternational Journal of TechnologyDwinaRahmayaniОценок пока нет
- Design Fabrication and Evaluation of A MДокумент92 страницыDesign Fabrication and Evaluation of A Mphillip chirongweОценок пока нет
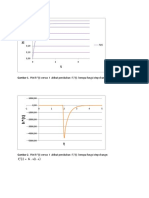
- T U - K T F: Gambar 1. Plot H (T) Versus T Akibat Perubahan FДокумент2 страницыT U - K T F: Gambar 1. Plot H (T) Versus T Akibat Perubahan FDwinaRahmayaniОценок пока нет
- 03 Secondary Wastewater TreatmentДокумент36 страниц03 Secondary Wastewater TreatmentDwinaRahmayaniОценок пока нет
- Review On The Design of A Tray Dryer System For Agricultural ProductsДокумент10 страницReview On The Design of A Tray Dryer System For Agricultural ProductsDwinaRahmayaniОценок пока нет
- Andaliman 1Документ8 страницAndaliman 1DwinaRahmayaniОценок пока нет
- Mutu Kertas Dari Pulp Batang Kelapa SawiДокумент12 страницMutu Kertas Dari Pulp Batang Kelapa SawiDwinaRahmayaniОценок пока нет
- Suggesting: AcceptingДокумент5 страницSuggesting: AcceptingDwinaRahmayaniОценок пока нет
- Koding Konsep Dasar Dan The King Ganesha Operation Smester 1 XII IPA. IntroДокумент12 страницKoding Konsep Dasar Dan The King Ganesha Operation Smester 1 XII IPA. IntroDwinaRahmayaniОценок пока нет
- 2016 0504 Data DiscriminationДокумент29 страниц2016 0504 Data DiscriminationDwinaRahmayaniОценок пока нет
- El Nadi2016Документ22 страницыEl Nadi2016DwinaRahmayaniОценок пока нет
- ATKДокумент16 страницATKDwinaRahmayaniОценок пока нет
- K20502en Ed6 Yardy Ev Yardy Duct en NTДокумент52 страницыK20502en Ed6 Yardy Ev Yardy Duct en NTUtzu SandruОценок пока нет
- Hyperco CatalogДокумент28 страницHyperco CatalogFallo SusiloОценок пока нет
- Cilindro Roemheld 1474Документ2 страницыCilindro Roemheld 1474AdrianaОценок пока нет
- Hydraulic Power Unit EДокумент8 страницHydraulic Power Unit EDipankar KhawasОценок пока нет
- 0.1 Hire Charges of Plants & Machinery: Basic RatesДокумент4 страницы0.1 Hire Charges of Plants & Machinery: Basic RatesSanjan SameerОценок пока нет
- Yamaha Nmax 155 - Safety Pre-Operation ChecksДокумент1 страницаYamaha Nmax 155 - Safety Pre-Operation Checksmotley crewzОценок пока нет
- Analysis of Balsa Wood Bridge ModelsДокумент28 страницAnalysis of Balsa Wood Bridge ModelsHazem EmamОценок пока нет
- Ropar ItemInventoryItemWiseList 10-02-2024Документ2 страницыRopar ItemInventoryItemWiseList 10-02-2024lahotishreyanshОценок пока нет
- Hidraulic Hammers Yekn0343Документ12 страницHidraulic Hammers Yekn0343Andy DellepianeОценок пока нет
- SG6250HV-MV Container InstallationДокумент3 страницыSG6250HV-MV Container InstallationJesica SantibañezОценок пока нет
- Simple Harmonic MotionДокумент102 страницыSimple Harmonic MotionYugandhar VeeramachaneniОценок пока нет
- BNP 20103 Hydraulic JumpДокумент4 страницыBNP 20103 Hydraulic JumpKomputershengalОценок пока нет
- Rule Bki V Rules For Materials 2009Документ249 страницRule Bki V Rules For Materials 2009Saadi AlimОценок пока нет
- AS2784Документ8 страницAS2784Randy Domingo FanerОценок пока нет
- 11 - 09 - Equippements Iacs 11 - 09 - Iacs Positioning EquipmentsДокумент2 страницы11 - 09 - Equippements Iacs 11 - 09 - Iacs Positioning EquipmentssxturboОценок пока нет
- Pump VP1 ParkerДокумент8 страницPump VP1 ParkerDian PramadiОценок пока нет
- Full Line Catalog: P-SeriesДокумент44 страницыFull Line Catalog: P-Seriesyoopr2Оценок пока нет
- Output 16 TO 1540 KW Pressure Jet Fuel Oil BurnersДокумент16 страницOutput 16 TO 1540 KW Pressure Jet Fuel Oil BurnersRawan Alwan ZarifОценок пока нет
- Leads - Hema1Документ12 страницLeads - Hema1Hema AnanthkumarОценок пока нет
- Muhammad Hanif Hidayat - 33120016 - LT2A - English Script PresentationДокумент2 страницыMuhammad Hanif Hidayat - 33120016 - LT2A - English Script PresentationMuhammad Hanif HidayatОценок пока нет