Вам также может понравиться
- Alcohol, Drugs and Medication in Pregnancy: The Long Term Outcome for the ChildОт EverandAlcohol, Drugs and Medication in Pregnancy: The Long Term Outcome for the ChildPhilip M PreeceРейтинг: 1 из 5 звезд1/5 (1)
- Option Com - Content&view Section&layout Blog&id 3 &itemid 60 FaqsДокумент7 страницOption Com - Content&view Section&layout Blog&id 3 &itemid 60 FaqsJig GamoloОценок пока нет
- Ven ListДокумент24 страницыVen ListWaf EtanoОценок пока нет
- Surgical Pathology of The Gastrointestinal System Bacterial, Fungal, Viral, and Parasitic Infections PDFДокумент234 страницыSurgical Pathology of The Gastrointestinal System Bacterial, Fungal, Viral, and Parasitic Infections PDFBogdan CarabasОценок пока нет
- SHARONДокумент65 страницSHARONFaith InstitutionsОценок пока нет
- Teleworking Risks and PreventionДокумент32 страницыTeleworking Risks and Preventionelena martinОценок пока нет
- Pa Tho Physiology of Hodgkin'sДокумент10 страницPa Tho Physiology of Hodgkin'sIvica Rae100% (1)
- NCP On Postpartum MotherДокумент9 страницNCP On Postpartum MotherM.S.H Tube100% (1)
- Pathophysiology of IBDДокумент11 страницPathophysiology of IBDOktarina Heni SunandarОценок пока нет
- 3 IClinicSys Presentation 2018 (As of August 2018)Документ21 страница3 IClinicSys Presentation 2018 (As of August 2018)desiree33% (3)
- Califmed00049 0003Документ7 страницCalifmed00049 0003Lola Khalid050Оценок пока нет
- Aspectos Clínicos Da Fenilcetonúria em Serviço de Referência em Triagem Neonatal Da BahiaДокумент6 страницAspectos Clínicos Da Fenilcetonúria em Serviço de Referência em Triagem Neonatal Da BahiaSara SaraОценок пока нет
- Childhood Eating Disorders British National Surveillance StudyДокумент7 страницChildhood Eating Disorders British National Surveillance StudygharrisanОценок пока нет
- Two-Step Process For ED UTI Screening in Febrile Young Children: Reducing Catheterization RatesДокумент8 страницTwo-Step Process For ED UTI Screening in Febrile Young Children: Reducing Catheterization RatesJean Pierre RojasОценок пока нет
- Recent Advances in Phenylketonuria ReviewДокумент12 страницRecent Advances in Phenylketonuria ReviewJesus Arturo VelazcoОценок пока нет
- 2518-Article Text-13816-1-10-20201215Документ6 страниц2518-Article Text-13816-1-10-20201215JAN CAMILLE LENONОценок пока нет
- An Overview of Newborn Screening: Paul A. Levy, MDДокумент10 страницAn Overview of Newborn Screening: Paul A. Levy, MDSardono WidinugrohoОценок пока нет
- Prenatal Screening SOGCДокумент13 страницPrenatal Screening SOGCBrendaОценок пока нет
- 1732-Article Text-27781-3-10-20220428Документ7 страниц1732-Article Text-27781-3-10-20220428Sarah Ong SiuОценок пока нет
- CAP - 11 Year Neurobehavioural OutcomesДокумент13 страницCAP - 11 Year Neurobehavioural OutcomesManju KumariОценок пока нет
- American Academy of Pediatrics Metabolic Disorders 2014 Practice TestДокумент43 страницыAmerican Academy of Pediatrics Metabolic Disorders 2014 Practice TestPrabu KumarОценок пока нет
- Jurnal AnakДокумент4 страницыJurnal AnakHumairah AnandaОценок пока нет
- A Study Protocol For The Prospective Validation Study Screening Programme For Pre-Eclampsia (SPREE)Документ20 страницA Study Protocol For The Prospective Validation Study Screening Programme For Pre-Eclampsia (SPREE)César GardeazábalОценок пока нет
- A Multiparity Woman With Pregnancy of Recurrent Anencephaly A Rare Case ReportДокумент2 страницыA Multiparity Woman With Pregnancy of Recurrent Anencephaly A Rare Case ReportInternational Journal of Innovative Science and Research TechnologyОценок пока нет
- Fenilcetonuria Genetica 2014Документ8 страницFenilcetonuria Genetica 2014Jhon NavarroОценок пока нет
- PrognosisДокумент7 страницPrognosispenyimpanankoas8Оценок пока нет
- A Retrospective Review of Newborn Screening For Congenital Hypothyroidism and Newborn Thyroid Disease at A Major Medical CenterДокумент8 страницA Retrospective Review of Newborn Screening For Congenital Hypothyroidism and Newborn Thyroid Disease at A Major Medical CenterJohn Paul Driz ViloriaОценок пока нет
- Mother's Knowledge and Attitude Towards Newborn ScreeningДокумент39 страницMother's Knowledge and Attitude Towards Newborn ScreeningNicky Jean100% (1)
- Tkachenko 2016Документ17 страницTkachenko 2016Phạm Văn HiệpОценок пока нет
- 16NBSHemoglobinDisorders IJNS2021Документ13 страниц16NBSHemoglobinDisorders IJNS2021JAN CAMILLE LENONОценок пока нет
- Invited Presentations and Presentations by Organisations and Societies / International Journal of Gynecology & Obstetrics 119S3 (2012) S161-S260Документ2 страницыInvited Presentations and Presentations by Organisations and Societies / International Journal of Gynecology & Obstetrics 119S3 (2012) S161-S260constantinoploqweОценок пока нет
- Performance of Fetal, Perinatal, and Pediatric AutopsiesДокумент6 страницPerformance of Fetal, Perinatal, and Pediatric AutopsiesbeosroОценок пока нет
- Clinical Review: Prevalence, Diagnosis, and Treatment of AnkyloglossiaДокумент7 страницClinical Review: Prevalence, Diagnosis, and Treatment of AnkyloglossiaisnaftyaОценок пока нет
- Reviewed - IJANS-Recent Antenatal Investigations For Better Pregnancy OutcomesДокумент4 страницыReviewed - IJANS-Recent Antenatal Investigations For Better Pregnancy Outcomesiaset123Оценок пока нет
- Deficiencia Intelectual - B6Документ7 страницDeficiencia Intelectual - B6fhenrique77Оценок пока нет
- Assessment of Mothers Care Toward Their Children Having PhenylketonuriaДокумент14 страницAssessment of Mothers Care Toward Their Children Having Phenylketonuriaأميرة خليلОценок пока нет
- Congenital Anomalies Parents Concerns and Opinions To - 2018 - European JournaДокумент1 страницаCongenital Anomalies Parents Concerns and Opinions To - 2018 - European Journaburak yolcuОценок пока нет
- Terminal Complement Activation in Preeclampsia - Burwick 2018Документ9 страницTerminal Complement Activation in Preeclampsia - Burwick 2018For GoogleОценок пока нет
- 33873-Article Text-121761-1-10-20170831Документ6 страниц33873-Article Text-121761-1-10-20170831AnggaОценок пока нет
- Intervencion Temprana SuccionДокумент13 страницIntervencion Temprana Succionjulianamd87Оценок пока нет
- Journal Edited Reyes FinalДокумент7 страницJournal Edited Reyes Finaljon paul reyesОценок пока нет
- Aspirin For Evidence-Based Preeclampsia Prevention Trial: Effect of Aspirin On Length of Stay in The Neonatal Intensive Care UnitДокумент6 страницAspirin For Evidence-Based Preeclampsia Prevention Trial: Effect of Aspirin On Length of Stay in The Neonatal Intensive Care UnitAripin Ari AripinОценок пока нет
- DR Kumar Ponnusamy Biochemistry Genetics USMLE Preparatory Course BIOGEN Reusable On Line Resources For Large Group Teaching Learning in Relatively SHДокумент2 страницыDR Kumar Ponnusamy Biochemistry Genetics USMLE Preparatory Course BIOGEN Reusable On Line Resources For Large Group Teaching Learning in Relatively SHPonnusamy KumarОценок пока нет
- Twinkle Patel SynopsisДокумент20 страницTwinkle Patel Synopsissa867069Оценок пока нет
- Cardiovascular Risk in Hypertension Open Question 13Документ2 страницыCardiovascular Risk in Hypertension Open Question 13Nurulhuda RashidОценок пока нет
- Abstract in Surgery PDFДокумент188 страницAbstract in Surgery PDFDrAmmar MagdyОценок пока нет
- ARTICUÑO Es enДокумент9 страницARTICUÑO Es enJUANITA MORENO OMEОценок пока нет
- Neonatal Palliative Care: Current Opinion in Pediatrics January 2017Документ7 страницNeonatal Palliative Care: Current Opinion in Pediatrics January 2017Raul VillacresОценок пока нет
- Post Partum Maternal Morbidities and TheДокумент102 страницыPost Partum Maternal Morbidities and TheBagusHibridaОценок пока нет
- General Gynaecology:, Leo Francis AquilizanДокумент15 страницGeneral Gynaecology:, Leo Francis AquilizanesjayemОценок пока нет
- Aspre TrialДокумент9 страницAspre TrialAryadna Bautista MarquezОценок пока нет
- Retrospective Analysis of Risk Factors For Low 1-Minute Apgar Scores in Term NeonatesДокумент10 страницRetrospective Analysis of Risk Factors For Low 1-Minute Apgar Scores in Term Neonatesputri vinia /ilove cuteОценок пока нет
- Background: ISRCTN00842661Документ17 страницBackground: ISRCTN00842661Sav GaОценок пока нет
- Phenylketonuria (PKU) : (Metabolic Condition: Amino Acid Disorder)Документ1 страницаPhenylketonuria (PKU) : (Metabolic Condition: Amino Acid Disorder)reriti2008Оценок пока нет
- Pku GRP 1Документ19 страницPku GRP 1Jersey MariОценок пока нет
- Neonatal Jaundice Literature ReviewДокумент6 страницNeonatal Jaundice Literature Reviewaflsmawld100% (1)
- Adverse Effects of Fetal Cocaine Exposure On Neonatal AuditoryДокумент9 страницAdverse Effects of Fetal Cocaine Exposure On Neonatal AuditoryFabiola Jacinto RubíОценок пока нет
- Level of Awareness of Post Partum Mothers On Newborn Screening EssaysДокумент4 страницыLevel of Awareness of Post Partum Mothers On Newborn Screening EssaysJig GamoloОценок пока нет
- MCH Group 8Документ12 страницMCH Group 8nafisatmuhammed452Оценок пока нет
- Jurnal KedokteranДокумент6 страницJurnal Kedokteranusk.ppdsobgynganjil2022Оценок пока нет
- Feeding Proem.xДокумент4 страницыFeeding Proem.xMuhammad Yusril JalilОценок пока нет
- 637Документ4 страницы637Sharuk AhamedОценок пока нет
- Journal PNC Asuhan Kebidanan Ibu Nifas Post SC Atas Indikasi Pre-Eklamsia Berat Di Ruangan Rawat Gabung Kebidanan Rsup Dr.M.DjamilДокумент12 страницJournal PNC Asuhan Kebidanan Ibu Nifas Post SC Atas Indikasi Pre-Eklamsia Berat Di Ruangan Rawat Gabung Kebidanan Rsup Dr.M.DjamilRizkya kurnia sidiqОценок пока нет
- Constipation: Beyond The Old ParadigmsДокумент18 страницConstipation: Beyond The Old ParadigmsJhonatan Efraín López CarbajalОценок пока нет
- 17-OHPC To Prevent Recurrent Preterm Birth in Singleton Gestations (PROLONG Study) : A Multicenter, International, Randomized Double-Blind TrialДокумент10 страниц17-OHPC To Prevent Recurrent Preterm Birth in Singleton Gestations (PROLONG Study) : A Multicenter, International, Randomized Double-Blind Trialomarmeftah838Оценок пока нет
- Protocol For The Prospective Validation Study: Screening Programme For Pre-Eclampsia' (SPREE)Документ5 страницProtocol For The Prospective Validation Study: Screening Programme For Pre-Eclampsia' (SPREE)Shelly MargarethaОценок пока нет
- Congenital Hyperinsulinism: A Practical Guide to Diagnosis and ManagementОт EverandCongenital Hyperinsulinism: A Practical Guide to Diagnosis and ManagementDiva D. De León-CrutchlowОценок пока нет
- Chiu 2019Документ8 страницChiu 2019Erika WulandariОценок пока нет
- Apexum in EndodonticsДокумент4 страницыApexum in EndodonticsAmit KumarОценок пока нет
- Untitled Document - EditedДокумент2 страницыUntitled Document - EditedBenjamin MuriithiОценок пока нет
- Jurnal SR 3Документ12 страницJurnal SR 3Zaina Khairunisa AsyaОценок пока нет
- Health-Care Workers' Occupational Exposures To Body Fluids in 21 Countries in Africa: Systematic Review and Meta-AnalysisДокумент17 страницHealth-Care Workers' Occupational Exposures To Body Fluids in 21 Countries in Africa: Systematic Review and Meta-AnalysisgusdeОценок пока нет
- Jurnal Kalsium PDFДокумент4 страницыJurnal Kalsium PDFAqbar S. P. PontohОценок пока нет
- PubLSIS II Survey Finding Report June 2018Документ620 страницPubLSIS II Survey Finding Report June 2018Mao udangОценок пока нет
- Contracted Pelvis and ManagementДокумент19 страницContracted Pelvis and Managementkirtyy20Оценок пока нет
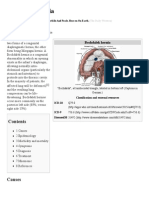
- Bochdalek Hernia - Wikipedia, The Free Encyclopedia PDFДокумент5 страницBochdalek Hernia - Wikipedia, The Free Encyclopedia PDFMilda InayahОценок пока нет
- Risk Assessment (Hirarc)Документ17 страницRisk Assessment (Hirarc)MNashОценок пока нет
- Labial Access For Lower TeethДокумент3 страницыLabial Access For Lower TeethHarish ChowdaryОценок пока нет
- Management of Allergic Rhinitis: P. Van Cauwenberge, H. Van HoeckeДокумент20 страницManagement of Allergic Rhinitis: P. Van Cauwenberge, H. Van HoeckePeter SalimОценок пока нет
- MI Consumer Guide To Health InsuranceДокумент40 страницMI Consumer Guide To Health InsuranceMichigan NewsОценок пока нет
- Module 6Документ22 страницыModule 6Jhoanna Lovely OntulanОценок пока нет
- 162 319 1 SMДокумент8 страниц162 319 1 SMAsniar RОценок пока нет
- Hca 600 Powerpoint Emr Plus Physician SectionДокумент20 страницHca 600 Powerpoint Emr Plus Physician Sectionapi-297065332Оценок пока нет
- NATIONAL INSURANCE - National Parivar Mediclaim PolicyДокумент31 страницаNATIONAL INSURANCE - National Parivar Mediclaim PolicyStigan IndiaОценок пока нет
- Disability Cert 15042019Документ8 страницDisability Cert 15042019Reshal AhmadОценок пока нет
- Inc Tnai IcnДокумент7 страницInc Tnai IcnDeena MelvinОценок пока нет
- IDS - Tuberculosis (Dr. Sy)Документ5 страницIDS - Tuberculosis (Dr. Sy)Renrenz PayumoОценок пока нет
- Radiation Safety Program: HDR Cobalt-60 and LDR Cesium-137 BRACHYTHERAPY FACILITYДокумент38 страницRadiation Safety Program: HDR Cobalt-60 and LDR Cesium-137 BRACHYTHERAPY FACILITYGregory CagampanОценок пока нет
- Physiotherapy Practices For Patients Undergoing Coronary Artery Bypass Grafting A Cross - Sectional StudyДокумент11 страницPhysiotherapy Practices For Patients Undergoing Coronary Artery Bypass Grafting A Cross - Sectional StudyhayaОценок пока нет
- Manejo Del Choque Séptico: Jesús Nicolás Hidalgo DelgadoДокумент40 страницManejo Del Choque Séptico: Jesús Nicolás Hidalgo DelgadoNicko HidalgoОценок пока нет