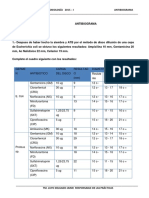
Вам также может понравиться
- Infertilidad MasculinaДокумент23 страницыInfertilidad MasculinaLuis Eduardo Soto GuardoОценок пока нет
- Preguntas para Resolver Tercer ParcialДокумент5 страницPreguntas para Resolver Tercer Parcialᴘᴜʀᴘʟᴇ vɪᴄОценок пока нет
- Leyes de La HerenciaДокумент6 страницLeyes de La Herencianayely.carpioОценок пока нет
- Temario de Biología y Genética II para El Segundo Parcial 2020Документ10 страницTemario de Biología y Genética II para El Segundo Parcial 2020majoОценок пока нет
- Etiología y Factores Que Intervienen en La EtiologíaДокумент13 страницEtiología y Factores Que Intervienen en La EtiologíaNhakinaОценок пока нет
- 2.1 Diferenciación Sexual NormalДокумент30 страниц2.1 Diferenciación Sexual Normalzenith44990% (1)
- 1 Parcial EmbriologiaДокумент9 страниц1 Parcial EmbriologiaAmada MagalinОценок пока нет
- Introducción A La Regulación y Señalización MolecularДокумент41 страницаIntroducción A La Regulación y Señalización MolecularMarina Itzel Castillo Ambriz50% (2)
- TRABAJO PRACTICO ENFERMEDADES CONGENITAS CalcutaДокумент15 страницTRABAJO PRACTICO ENFERMEDADES CONGENITAS CalcutaGerardo PaneloОценок пока нет
- Capitulo 9. Genetica.Документ49 страницCapitulo 9. Genetica.Mauricio GodoyОценок пока нет
- Desarrollo AnimalДокумент16 страницDesarrollo AnimalLaura AguilarОценок пока нет
- C2 Introduccion y TerminologiaДокумент16 страницC2 Introduccion y TerminologiaCesar AriasОценок пока нет
- Hermafroditismo 05Документ18 страницHermafroditismo 05Antonio AspireОценок пока нет
- GENETICAДокумент25 страницGENETICAJhamil LopezОценок пока нет
- Qué Es La EspermatogénesisДокумент5 страницQué Es La EspermatogénesisCarola Garrido100% (1)
- Resumen PSU Biologia PDVДокумент28 страницResumen PSU Biologia PDVHumberto Bustos CalabaceroОценок пока нет
- Nombre Del Profesor: - Ricardo Ortega PérezДокумент4 страницыNombre Del Profesor: - Ricardo Ortega PérezIcela VAОценок пока нет
- Toxicologia Del Aparato ReproductorДокумент7 страницToxicologia Del Aparato ReproductorJesus Raul Torres IbarraОценок пока нет
- Reproducci N y Desarrollo-Cristi N Delgado 1 - 2015Документ20 страницReproducci N y Desarrollo-Cristi N Delgado 1 - 2015Bruno VargasОценок пока нет
- Terminología GenéticaДокумент10 страницTerminología GenéticaAndrea BerlianОценок пока нет
- Resumen de Embriología Regulacion MolecularДокумент5 страницResumen de Embriología Regulacion MolecularJeisson Jauregui100% (1)
- Trabajo de Investigacion de BiologiaДокумент10 страницTrabajo de Investigacion de Biologiacavernicola0050% (2)
- Drosophila MelanogasterДокумент9 страницDrosophila MelanogastersudroleyduОценок пока нет
- Malformaciones Congénitas-1Документ10 страницMalformaciones Congénitas-1AlanRCОценок пока нет
- Resumen EmbriologíaДокумент4 страницыResumen EmbriologíaYonalí MármolОценок пока нет
- Histologia Aparato Reproductor FemeninoДокумент2 страницыHistologia Aparato Reproductor FemeninoLuis EsparzaОценок пока нет
- Capitulo 3 Formacióndeunanuevavida PDFДокумент3 страницыCapitulo 3 Formacióndeunanuevavida PDFAinat Ollitsac ZurcОценок пока нет
- Trastornos Diferenciacion SexualДокумент9 страницTrastornos Diferenciacion SexualLuciaGomezОценок пока нет
- Embrio RepasoooДокумент14 страницEmbrio RepasoooReyes Ivan García CuevasОценок пока нет
- Guía de Estudio Tercer Parcial Biología HumanaДокумент13 страницGuía de Estudio Tercer Parcial Biología Humanakevincantillano33Оценок пока нет
- Investigación de La Reproducción CélularДокумент28 страницInvestigación de La Reproducción CélularJacqueline FranciscoОценок пока нет
- Alteraciones Embrio Facmed 2018 1Документ41 страницаAlteraciones Embrio Facmed 2018 1Emiliano SolísОценок пока нет
- AnomaliasДокумент5 страницAnomaliasVanessa Rodriguez OlmosОценок пока нет
- Cromosoma - Hans ViscarraДокумент8 страницCromosoma - Hans ViscarraJuan ViscarraОценок пока нет
- Ensayo Biologia Del Desarrollo AnimalДокумент19 страницEnsayo Biologia Del Desarrollo AnimalNADIAОценок пока нет
- Capitulo 2 - Patogenia Del CancerДокумент7 страницCapitulo 2 - Patogenia Del Cancerariadna_maciasОценок пока нет
- Parte 2: Bases Moleculares Del Cáncer Muy Controlada de Eventos. Estos Eventos Dependen de Niveles Adecuados de Transcripción y Traducción deДокумент3 страницыParte 2: Bases Moleculares Del Cáncer Muy Controlada de Eventos. Estos Eventos Dependen de Niveles Adecuados de Transcripción y Traducción deMaria JoseОценок пока нет
- Cuestionario 3 - DesarrolloДокумент3 страницыCuestionario 3 - DesarrolloLeonardo Francis Yaranga MoisésОценок пока нет
- Guia Morfologia 1Документ11 страницGuia Morfologia 1Ana murilloОценок пока нет
- Glosario de TérminosДокумент8 страницGlosario de Términosluci mezaОценок пока нет
- Plasmidos de MolecularДокумент7 страницPlasmidos de Molecularandres zuluagaОценок пока нет
- GenéticaДокумент9 страницGenéticaSharon VenturaОценок пока нет
- Unidad 2 MATERNO INFANTIL GeneticaДокумент32 страницыUnidad 2 MATERNO INFANTIL GeneticaPame MedranoОценок пока нет
- Ensayo Mutacion y Cariotipo Juan OñateДокумент5 страницEnsayo Mutacion y Cariotipo Juan Oñateyulibeth pereira mielessОценок пока нет
- Informe Celulas SexualesДокумент17 страницInforme Celulas Sexualesfran asanzaОценок пока нет
- Ud6 - Citogenética y CancerДокумент9 страницUd6 - Citogenética y Cancerarino.martinez.monicaaОценок пока нет
- Articulo 4Документ5 страницArticulo 4mavix726Оценок пока нет
- Act - 3.3 - Kizimia - Fantini - Resumen. Sexo y CerebroДокумент6 страницAct - 3.3 - Kizimia - Fantini - Resumen. Sexo y CerebroRafa FanОценок пока нет
- ESPERMATOGENESISДокумент23 страницыESPERMATOGENESISMARTÍNEZ CONTIZ LENY YAMILEОценок пока нет
- Preguntas A Examen de GeneticaДокумент3 страницыPreguntas A Examen de GeneticaMarian Orte SáenzОценок пока нет
- ONCOGENESДокумент93 страницыONCOGENESMarco Herrera Flores100% (1)
- 003 Informe 16 BiologiaДокумент2 страницы003 Informe 16 BiologiaJORDY SMITH CORNEJO FLORESОценок пока нет
- La MutacionДокумент4 страницыLa Mutacion0607195Оценок пока нет
- Unidad 2 Materno InfantilДокумент32 страницыUnidad 2 Materno InfantilGloria SeibertОценок пока нет
- Fallas en La MeiosisДокумент4 страницыFallas en La MeiosisEduardo Torres Palacios100% (3)
- Glosario de Términos GenéticosДокумент4 страницыGlosario de Términos GenéticosJhunhely Zantos100% (1)
- CAPÍTULO 9 - Gametogénesis y FecundaciónДокумент48 страницCAPÍTULO 9 - Gametogénesis y FecundaciónSandra milena Diaz mercadoОценок пока нет
- Ambiguedad GenitalДокумент16 страницAmbiguedad GenitalKowe xDОценок пока нет
- Hipogonadismo ExpoДокумент21 страницаHipogonadismo ExpoMelanie Bravo DavidОценок пока нет
- Anexo 1Документ2 страницыAnexo 1Brandon Chahua ObregonОценок пока нет
- Anexo 1Документ2 страницыAnexo 1Brandon Chahua ObregonОценок пока нет
- Juan SociodramaДокумент1 страницаJuan SociodramaBrandon Chahua ObregonОценок пока нет
- Resumen XDДокумент5 страницResumen XDBrandon Chahua ObregonОценок пока нет
- PRÁCTICAДокумент1 страницаPRÁCTICABrandon Chahua ObregonОценок пока нет
- ECOLOGIAДокумент2 страницыECOLOGIABrandon Chahua Obregon0% (1)
- Resumen 1Документ4 страницыResumen 1Brandon Chahua ObregonОценок пока нет
- Liderazgo en Medicina - MedigraphicДокумент9 страницLiderazgo en Medicina - MedigraphicBrandon Chahua ObregonОценок пока нет
- 01 SegmentosДокумент3 страницы01 SegmentosBrandon Chahua ObregonОценок пока нет
- Liderazgo en Medicina - MedigraphicДокумент9 страницLiderazgo en Medicina - MedigraphicBrandon Chahua ObregonОценок пока нет
- Dialnet CalidadYLiderazgoEnMedicina 4701391 PDFДокумент11 страницDialnet CalidadYLiderazgoEnMedicina 4701391 PDFJuarez 3710Оценок пока нет
- INDICEДокумент2 страницыINDICEBrandon Chahua ObregonОценок пока нет
- Bienvenido PDFДокумент5 страницBienvenido PDFRaquel Pérez PérezОценок пока нет
- CRONOGRAMAДокумент1 страницаCRONOGRAMABrandon Chahua ObregonОценок пока нет
- Conclusion EsДокумент2 страницыConclusion EsBrandon Chahua Obregon100% (1)
- PLANTILLA-DE-EXPOSICION BrandonДокумент5 страницPLANTILLA-DE-EXPOSICION BrandonBrandon Chahua ObregonОценок пока нет
- PLANTILLA-DE-EXPOSICION BrandonДокумент5 страницPLANTILLA-DE-EXPOSICION BrandonBrandon Chahua ObregonОценок пока нет
- 4tl4s Its MedicinafullДокумент92 страницы4tl4s Its MedicinafullCézhar Navarro Pardavé100% (1)
- Malla Curricular - XLДокумент4 страницыMalla Curricular - XLCANAL DE ING. ELECTRONICA UNJFSCОценок пока нет
- Anquilostiomosis 2019Документ12 страницAnquilostiomosis 2019Brandon Chahua ObregonОценок пока нет
- Resultados 1Документ1 страницаResultados 1Brandon Chahua ObregonОценок пока нет
- Cuestionario #07Документ2 страницыCuestionario #07Brandon Chahua ObregonОценок пока нет
- Cuestionario #05 AntibiogramaДокумент5 страницCuestionario #05 AntibiogramaBrandon Chahua ObregonОценок пока нет
- Cy A 4to Primara La CélulaДокумент1 страницаCy A 4to Primara La CélulaBrandon Chahua ObregonОценок пока нет
- Cuestionario 111111Документ6 страницCuestionario 111111Brandon Chahua ObregonОценок пока нет
- Qué Es Una EnzimaДокумент6 страницQué Es Una EnzimaBrandon Chahua Obregon100% (1)
- Qué Es Una EnzimaДокумент30 страницQué Es Una EnzimaBrandon Chahua ObregonОценок пока нет
- Bases Moleculares de La FecundacionДокумент11 страницBases Moleculares de La FecundacionBrandon Chahua ObregonОценок пока нет
- Monografía de La LepraДокумент23 страницыMonografía de La Leprasamcr000100% (4)
- Cuestionario N 9Документ5 страницCuestionario N 9Brandon Chahua ObregonОценок пока нет
- Revista 21 1 2014 4Документ10 страницRevista 21 1 2014 4Maria Peschiera BenitesОценок пока нет
- Strongyloides Stercoralis EN ESCOLARES DE LA UNIDAD EDUCATIVA BOLIVARIANA GUAIMIRE, GUAIMIRE, ESTADO BOLÍVAR.Документ64 страницыStrongyloides Stercoralis EN ESCOLARES DE LA UNIDAD EDUCATIVA BOLIVARIANA GUAIMIRE, GUAIMIRE, ESTADO BOLÍVAR.Ivor Enrique Osorio MoralesОценок пока нет
- Práctica 11 - Dilatación de SólidosДокумент3 страницыPráctica 11 - Dilatación de SólidosSAUL BOLIVAR SARMIENTO CARCHIОценок пока нет
- Plan de Acción Comunitario Leg TabacayДокумент4 страницыPlan de Acción Comunitario Leg TabacayGabriel EspinozaОценок пока нет
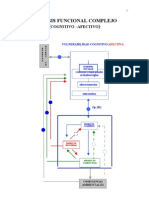
- Walter Riso - Análisis Funcional ComplejoДокумент8 страницWalter Riso - Análisis Funcional ComplejoSamantha Rojas GuzmanОценок пока нет
- Tarea 5.1 ETICA Y GENETICAДокумент2 страницыTarea 5.1 ETICA Y GENETICALuis GarciasОценок пока нет
- Que Es Un EquimosisДокумент4 страницыQue Es Un Equimosisgreym garciaОценок пока нет
- Trabajo LubricacionДокумент10 страницTrabajo Lubricacionjuanes estebanОценок пока нет
- La Neurociencia en El Sistema JudicialДокумент6 страницLa Neurociencia en El Sistema JudicialJosé María ValmisaОценок пока нет
- Control Ambiental Del Relleno Sanitario Los AngelesДокумент8 страницControl Ambiental Del Relleno Sanitario Los AngelesJhoan Sebastian PerezОценок пока нет
- PiromaníaДокумент7 страницPiromaníaAnthony MacbrayanОценок пока нет
- FILA A Prueba Nomenclatura InorganicaДокумент5 страницFILA A Prueba Nomenclatura InorganicaANA MARIA MARIHUAN100% (1)
- Relaciones IntermaxilaresДокумент6 страницRelaciones IntermaxilarescelesteОценок пока нет
- Cáncer de PulmónДокумент17 страницCáncer de PulmónPatricia Morales SepúlvedaОценок пока нет
- Proyecto Trabajo Social-Diana Vargas Modificado - NewДокумент17 страницProyecto Trabajo Social-Diana Vargas Modificado - NewYorman BaccaОценок пока нет
- Investigacion DocumentalДокумент6 страницInvestigacion DocumentalkarinaОценок пока нет
- Material Informativo 9.1Документ4 страницыMaterial Informativo 9.1Pedro Jacobo FernándezОценок пока нет
- Caso Práctico Higiene 4Документ4 страницыCaso Práctico Higiene 4Victor RodrigesОценок пока нет
- 7 - Medición de Temperatura (Pirometros) .Документ5 страниц7 - Medición de Temperatura (Pirometros) .Leonel SittnerОценок пока нет
- Historia de La Gastronomia MundialДокумент4 страницыHistoria de La Gastronomia Mundialrebeca velasque guerraОценок пока нет
- Los 7 Mejores Alimentos para Subir La HemoglobinaДокумент2 страницыLos 7 Mejores Alimentos para Subir La HemoglobinaNancy EscuderoОценок пока нет
- 10 Open PitДокумент79 страниц10 Open PitJAVIER PATRICIO DE JESÚS LAYANA LÓPEZОценок пока нет
- Cultos Innombrables FICHA EDITABLEДокумент1 страницаCultos Innombrables FICHA EDITABLEOtra cuenta sin sentidoОценок пока нет
- Semiología UrológicaДокумент60 страницSemiología UrológicaScott WebbОценок пока нет
- TDR 10 Implementos de Seguridad IIДокумент3 страницыTDR 10 Implementos de Seguridad IIlordevilsОценок пока нет
- Desarrollor Ejercicio 1 UNIDAD 1 de GanaciasДокумент6 страницDesarrollor Ejercicio 1 UNIDAD 1 de Ganaciasraul chirinosОценок пока нет
- Carta Responsiva, Afiliación y Compromiso ADEMEBA ChiapasДокумент6 страницCarta Responsiva, Afiliación y Compromiso ADEMEBA ChiapasDariana CoutiñoОценок пока нет
- Hoja Datos y Seguridad Quimico W200 - ES 2015Документ11 страницHoja Datos y Seguridad Quimico W200 - ES 2015Alejandra Lorena Rojas GarzónОценок пока нет
- Enfeermedades Del Sistema RespiratorioДокумент15 страницEnfeermedades Del Sistema RespiratorioMabel RomaniОценок пока нет
- Constitucion de EmpresaДокумент3 страницыConstitucion de EmpresaRalf CardenasОценок пока нет
- El Monje Que Vendio Su Ferrari: Una Fábula EspiritualОт EverandEl Monje Que Vendio Su Ferrari: Una Fábula EspiritualРейтинг: 4.5 из 5 звезд4.5/5 (1698)
- Tus Zonas Erroneas: Guía Para Combatir las Causas de la InfelicidadОт EverandTus Zonas Erroneas: Guía Para Combatir las Causas de la InfelicidadРейтинг: 4.5 из 5 звезд4.5/5 (1831)
- Psicología oscura: Una guía esencial de persuasión, manipulación, engaño, control mental, negociación, conducta humana, PNL y guerra psicológicaОт EverandPsicología oscura: Una guía esencial de persuasión, manipulación, engaño, control mental, negociación, conducta humana, PNL y guerra psicológicaРейтинг: 4.5 из 5 звезд4.5/5 (766)
- Tu cerebro emocional: Saca partido de lo que sientes y transforma tu vidaОт EverandTu cerebro emocional: Saca partido de lo que sientes y transforma tu vidaРейтинг: 5 из 5 звезд5/5 (2)
- Cómo hacer que te pasen cosas buenas: Entiende tu cerebro, gestiona tus emociones, mejora tu vidaОт EverandCómo hacer que te pasen cosas buenas: Entiende tu cerebro, gestiona tus emociones, mejora tu vidaРейтинг: 5 из 5 звезд5/5 (1870)
- La revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaОт EverandLa revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaРейтинг: 5 из 5 звезд5/5 (201)
- Yo Pude, ¡Tú Puedes!: Cómo tomar el control de tu bienestar emocional y convertirte en una persona imparable (edición revisada y expandida)От EverandYo Pude, ¡Tú Puedes!: Cómo tomar el control de tu bienestar emocional y convertirte en una persona imparable (edición revisada y expandida)Рейтинг: 5 из 5 звезд5/5 (7)
- Cómo Acertar en tu Nueva Relación de Pareja: Todas las claves para saber si tu relación tiene futuro... o te conviene salir corriendo de ahí.От EverandCómo Acertar en tu Nueva Relación de Pareja: Todas las claves para saber si tu relación tiene futuro... o te conviene salir corriendo de ahí.Рейтинг: 4.5 из 5 звезд4.5/5 (7)
- Resetea tu mente. Descubre de lo que eres capazОт EverandResetea tu mente. Descubre de lo que eres capazРейтинг: 5 из 5 звезд5/5 (196)
- Los Secretos De La Mente Millonaria: Domina el juego de la riquezaОт EverandLos Secretos De La Mente Millonaria: Domina el juego de la riquezaРейтинг: 5 из 5 звезд5/5 (457)
- ¡Tómate un respiro! Mindfulness: El arte de mantener la calma en medio de la tempestadОт Everand¡Tómate un respiro! Mindfulness: El arte de mantener la calma en medio de la tempestadРейтинг: 5 из 5 звезд5/5 (198)
- ¿Por qué mis padres no me aman?: Empezando a sanarОт Everand¿Por qué mis padres no me aman?: Empezando a sanarРейтинг: 4.5 из 5 звезд4.5/5 (33)
- Resumen de Pensar rápido pensar despacio de Daniel KahnemanОт EverandResumen de Pensar rápido pensar despacio de Daniel KahnemanРейтинг: 4.5 из 5 звезд4.5/5 (64)
- Homo antecessor: El nacimiento de una especieОт EverandHomo antecessor: El nacimiento de una especieРейтинг: 5 из 5 звезд5/5 (1)
- Mindfulness para principiantes: Medita sin meditarОт EverandMindfulness para principiantes: Medita sin meditarРейтинг: 5 из 5 звезд5/5 (53)
- Disciplina Mental: Técnicas infalibles para lograr todo lo que te propones y eliminar la pereza y la procrastinación de tu vida para siempreОт EverandDisciplina Mental: Técnicas infalibles para lograr todo lo que te propones y eliminar la pereza y la procrastinación de tu vida para siempreРейтинг: 5 из 5 звезд5/5 (3)
- El poder del optimismo: Herramientas para vivir de forma más positivaОт EverandEl poder del optimismo: Herramientas para vivir de forma más positivaРейтинг: 5 из 5 звезд5/5 (16)
- Influencia. La psicología de la persuasiónОт EverandInfluencia. La psicología de la persuasiónРейтинг: 4.5 из 5 звезд4.5/5 (14)
- Grimorio: Cómo lanzar y elaborar hechizos mágicos, aprender las prácticas wiccanas y desvelar los secretos de la brujería a través de un diario ritualОт EverandGrimorio: Cómo lanzar y elaborar hechizos mágicos, aprender las prácticas wiccanas y desvelar los secretos de la brujería a través de un diario ritualОценок пока нет
- Cómo terminar lo que empiezas: El arte de perseverar, pasar a la acción, ejecutar los planes y tener disciplinaОт EverandCómo terminar lo que empiezas: El arte de perseverar, pasar a la acción, ejecutar los planes y tener disciplinaРейтинг: 4.5 из 5 звезд4.5/5 (6)
- Teoría polivagal práctica y terapiaОт EverandTeoría polivagal práctica y terapiaРейтинг: 5 из 5 звезд5/5 (4)