Вам также может понравиться
- Systemic Lupus Erythematosus PathophysiologyДокумент8 страницSystemic Lupus Erythematosus Pathophysiologykathy92% (24)
- SLE Patogenesis & Patofisiologi PDFДокумент8 страницSLE Patogenesis & Patofisiologi PDFRIZQI IRFANSYAH100% (1)
- Designing and Drawing PropellerДокумент4 страницыDesigning and Drawing Propellercumpio425428100% (1)
- Pathophysiology Acute Bacterial MeningitisДокумент2 страницыPathophysiology Acute Bacterial MeningitisNadira Farah PrayogoОценок пока нет
- Mathematics - Mathematics of Magic - A Study in Probability, Statistics, Strategy and Game Theory XДокумент32 страницыMathematics - Mathematics of Magic - A Study in Probability, Statistics, Strategy and Game Theory XHarish HandОценок пока нет
- Systemic Lupus Erythematosus (SLE) : Genetic Factors Environmental FactorsДокумент5 страницSystemic Lupus Erythematosus (SLE) : Genetic Factors Environmental Factorsjoyrena ochondraОценок пока нет
- Pathophysiology - Rheumatoid ArthritisДокумент1 страницаPathophysiology - Rheumatoid ArthritisAngel FiloteoОценок пока нет
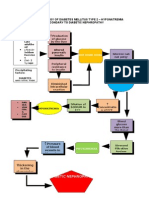
- Pathophysiology of Diabetes Mellitus Type 2Документ7 страницPathophysiology of Diabetes Mellitus Type 2arbyjamesОценок пока нет
- Marketing Micro and Macro EnvironmentДокумент8 страницMarketing Micro and Macro EnvironmentSumit Acharya100% (1)
- Group 3 Pathophysiology-of-BREAST-CANCERДокумент1 страницаGroup 3 Pathophysiology-of-BREAST-CANCERArisa VijungcoОценок пока нет
- Systemic Lupus Erythematosus Actual PathophysiologyДокумент1 страницаSystemic Lupus Erythematosus Actual PathophysiologyGhan Maria100% (2)
- Pathophysiology of Acute Kidney InjuryДокумент4 страницыPathophysiology of Acute Kidney InjuryJane Arian Berzabal0% (1)
- Pathophysiology of SLEДокумент16 страницPathophysiology of SLESeff CausapinОценок пока нет
- Pa Tho Physiology Sle, CompДокумент5 страницPa Tho Physiology Sle, CompHassan Bj MarabongОценок пока нет
- Qtsoi Concept MapДокумент5 страницQtsoi Concept MapGenella BabantoОценок пока нет
- Pathophysiology of Diabetes Mellitus Type 2Документ1 страницаPathophysiology of Diabetes Mellitus Type 2faula rocamora100% (3)
- Low Voltage Switchgear Specification: 1. ScopeДокумент6 страницLow Voltage Switchgear Specification: 1. ScopejendrikoОценок пока нет
- BSH 7005-15Документ129 страницBSH 7005-15Mark InnesОценок пока нет
- Pathophysiology of Leptospirosis and Dengue FeverДокумент5 страницPathophysiology of Leptospirosis and Dengue FeverKenneth Lagman100% (1)
- SLE PathophysiologyДокумент3 страницыSLE PathophysiologyRanela Kwinkee Pastor Salazar100% (7)
- Sle Concept Map Part 1Документ1 страницаSle Concept Map Part 1Vane UcatОценок пока нет
- Pathophysiology: Precipitating FactorДокумент6 страницPathophysiology: Precipitating FactorMark Anthony YabresОценок пока нет
- Pathophysiology in Liver CirrhosisДокумент4 страницыPathophysiology in Liver CirrhosisCyrus Ortalla RobinОценок пока нет
- Pathophysiology of Rheumatoid ArthritisДокумент1 страницаPathophysiology of Rheumatoid ArthritisGerardeanne ReposarОценок пока нет
- Patho of Pott's DiseaseДокумент2 страницыPatho of Pott's DiseaseIris Balino100% (1)
- Pathophysiology of CholeraДокумент1 страницаPathophysiology of CholeraFarr Krizha Tangkusan67% (3)
- Pathophysiology DMДокумент1 страницаPathophysiology DMMJ AmarilloОценок пока нет
- COPD PathophysiologyДокумент1 страницаCOPD Pathophysiologyaj ajОценок пока нет
- Diagram of Pathophysiology CancerДокумент5 страницDiagram of Pathophysiology CancerKristaMaeC.Lazo0% (3)
- Infectious Mononucleosis Concept MapДокумент4 страницыInfectious Mononucleosis Concept MapElle100% (1)
- Dengue Hemorrhagic Fever PathophysiologyДокумент4 страницыDengue Hemorrhagic Fever PathophysiologyKirk Espanol BigstoneОценок пока нет
- Systemic Lupus Erythematosus PathophysiologyДокумент8 страницSystemic Lupus Erythematosus PathophysiologySharmaineTaguitagOmli100% (1)
- Pathophysiology Acute PyelonephritisДокумент2 страницыPathophysiology Acute PyelonephritisAnonymous 75TDy2y100% (1)
- Pathophysiology of Hyperthyroidism and Thyroid StormДокумент3 страницыPathophysiology of Hyperthyroidism and Thyroid StormPen MontanteОценок пока нет
- POTTs Disease PathoДокумент3 страницыPOTTs Disease PathoEdgel QuidolesОценок пока нет
- Guillain Barre Syndrome PathophysiologyДокумент4 страницыGuillain Barre Syndrome Pathophysiologykathy100% (13)
- PathoConceptMap AIDSДокумент3 страницыPathoConceptMap AIDSKristen Babauta50% (2)
- 160kW SOFT STARTER - TAP HOLE 1Документ20 страниц160kW SOFT STARTER - TAP HOLE 1Ankit Uttam0% (1)
- Anemia PathophysiologyДокумент2 страницыAnemia PathophysiologyHoney Lorie D. Simbajon67% (6)
- Measles PathoДокумент1 страницаMeasles PathoAin JamelaОценок пока нет
- Acute Lymphoblastic Leukemia Pathophysiology: Precipitating Factors: Etiology: Predisposing FactorsДокумент3 страницыAcute Lymphoblastic Leukemia Pathophysiology: Precipitating Factors: Etiology: Predisposing FactorsromelynОценок пока нет
- Pa Tho Physiology Part 1Документ1 страницаPa Tho Physiology Part 1anonymous89ify100% (2)
- Systemic Lupus Erythematosus PathophysiologyДокумент8 страницSystemic Lupus Erythematosus PathophysiologyAnonymous OU6w8lX9Оценок пока нет
- Dengue PathophysiologyДокумент1 страницаDengue PathophysiologyRafael Miguel Alon Protacio50% (2)
- Aplastic Anemia Pa Tho PhysiologyДокумент3 страницыAplastic Anemia Pa Tho Physiologyneil052275% (4)
- Pathophysiology and Schematic Diagram of Typhoid FeverДокумент3 страницыPathophysiology and Schematic Diagram of Typhoid FeverCyrus De AsisОценок пока нет
- ALL PathophysiologyДокумент2 страницыALL PathophysiologyDeo Michael Rivera LlamasОценок пока нет
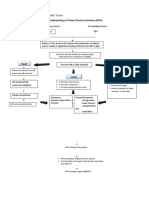
- Pathophysiology of Patent Ductus Arteroisus (PDA)Документ2 страницыPathophysiology of Patent Ductus Arteroisus (PDA)Rodel Yacas100% (1)
- Pa Tho Physiology of Open FractureДокумент2 страницыPa Tho Physiology of Open FracturegiffersonbОценок пока нет
- DB13 - Pathophysiology of AtherosclerosisДокумент2 страницыDB13 - Pathophysiology of Atherosclerosisi_vhie03Оценок пока нет
- Pathophysiology of LeukemiaДокумент1 страницаPathophysiology of LeukemiaCandra Dwipayana HamdinОценок пока нет
- Leukemia With PathophysiologyДокумент34 страницыLeukemia With Pathophysiologymabec pagaduan90% (41)
- Acute Lymphoblastic Leukemia (All)Документ35 страницAcute Lymphoblastic Leukemia (All)Mohamed RehanОценок пока нет
- Pathophysiology of Osteoporosis: AgingДокумент1 страницаPathophysiology of Osteoporosis: Agingnek100% (1)
- Systemic Lupus ErythematosusДокумент13 страницSystemic Lupus ErythematosusGrace RoselinyОценок пока нет
- Pathophysiology of Nephrotic SyndromeДокумент1 страницаPathophysiology of Nephrotic SyndromeRan MaОценок пока нет
- SLE PathophysiologyДокумент3 страницыSLE PathophysiologyyasiraОценок пока нет
- Cancer Pathophysiology To Be EditedДокумент5 страницCancer Pathophysiology To Be EditedEyySiEffVeeОценок пока нет
- Systemic Lupus ErythematosusДокумент37 страницSystemic Lupus ErythematosusFirman Ichlasul AmalОценок пока нет
- Pa Tho Physiology of DengueДокумент1 страницаPa Tho Physiology of Denguesinister17100% (1)
- PathophysiologyДокумент1 страницаPathophysiologynitlihpОценок пока нет
- Pathophysiology: HIV Infection and AIDSДокумент7 страницPathophysiology: HIV Infection and AIDSmeylin SОценок пока нет
- Acute Gout Case StudyДокумент2 страницыAcute Gout Case StudyElisa KerrОценок пока нет
- Pathophysiology of Hypersensitivity Type IIДокумент2 страницыPathophysiology of Hypersensitivity Type IItwin_smartyОценок пока нет
- Systemic Lupus Erythematous: Precipitating Factors: Environmental Drug-Induced InfectionДокумент10 страницSystemic Lupus Erythematous: Precipitating Factors: Environmental Drug-Induced Infectioninah krizia lagueОценок пока нет
- Valmed PathoДокумент1 страницаValmed PathoJez RarangОценок пока нет
- Med 1.11 - SleДокумент5 страницMed 1.11 - SleZazaОценок пока нет
- Module LESДокумент31 страницаModule LESclaudiaОценок пока нет
- MATH 181-Oblique Triangles and Vectors (11) Term 0306Документ16 страницMATH 181-Oblique Triangles and Vectors (11) Term 0306SharmaineTaguitagOmliОценок пока нет
- 8451Документ24 страницы8451tomzki007Оценок пока нет
- Module LESДокумент31 страницаModule LESclaudiaОценок пока нет
- CH01 FinalДокумент14 страницCH01 FinalSharmaineTaguitagOmliОценок пока нет
- Prevalence of Hypertension, Obesity, Diabetes and Smoking in Filipino AdultsДокумент63 страницыPrevalence of Hypertension, Obesity, Diabetes and Smoking in Filipino AdultsSharmaineTaguitagOmliОценок пока нет
- LKKДокумент14 страницLKKCandace WagnerОценок пока нет
- Triangle Congruence Proofs - Extra PracticeДокумент2 страницыTriangle Congruence Proofs - Extra PracticeSharmaineTaguitagOmliОценок пока нет
- Creative Commons Attribution-Noncommercial-Sharealike LicenseДокумент19 страницCreative Commons Attribution-Noncommercial-Sharealike LicenseSharmaineTaguitagOmliОценок пока нет
- 1 s2.0 S2214999615012643 MainДокумент7 страниц1 s2.0 S2214999615012643 MainTin BabistaОценок пока нет
- SavannahHarbor5R Restoration Plan 11 10 2015Документ119 страницSavannahHarbor5R Restoration Plan 11 10 2015siamak dadashzadeОценок пока нет
- Measurement and Scaling Techniques1Документ42 страницыMeasurement and Scaling Techniques1Ankush ChaudharyОценок пока нет
- Brosur YSIO X.preeДокумент20 страницBrosur YSIO X.preeRadiologi RSUD KilisuciОценок пока нет
- CUBE Dealer Book 2009Документ280 страницCUBE Dealer Book 2009maikruetzОценок пока нет
- The Body Shop Case Analysis. The Challenges of Managing Business As Holistic ConfigurationДокумент28 страницThe Body Shop Case Analysis. The Challenges of Managing Business As Holistic ConfigurationHanna AbejoОценок пока нет
- Multimedia System DesignДокумент95 страницMultimedia System DesignRishi Aeri100% (1)
- Roles and Responsibilities of An InstructorДокумент4 страницыRoles and Responsibilities of An InstructorMohanlal SainiОценок пока нет
- Prognostic Factors and Management of Patients With Choanal AtresiaДокумент7 страницPrognostic Factors and Management of Patients With Choanal Atresiafarah maulida martaОценок пока нет
- The Reason: B. FlowsДокумент4 страницыThe Reason: B. FlowsAryanti UrsullahОценок пока нет
- 105 2Документ17 страниц105 2Diego TobrОценок пока нет
- Paul Wade - The Ultimate Isometrics Manual - Building Maximum Strength and Conditioning With Static Training-Dragon Door Publications (2020) - 120-146Документ27 страницPaul Wade - The Ultimate Isometrics Manual - Building Maximum Strength and Conditioning With Static Training-Dragon Door Publications (2020) - 120-146usman azharОценок пока нет
- Term Paper Inorganic PolymersДокумент24 страницыTerm Paper Inorganic PolymersCasey Karua0% (1)
- Possessive Determiners: A. 1. A) B) C) 2. A) B) C) 3. A) B) C) 4. A) B) C) 5. A) B) C) 6. A) B) C) 7. A) B) C)Документ1 страницаPossessive Determiners: A. 1. A) B) C) 2. A) B) C) 3. A) B) C) 4. A) B) C) 5. A) B) C) 6. A) B) C) 7. A) B) C)Manuela Marques100% (1)
- Test Bank For The Psychology of Health and Health Care A Canadian Perspective 5th EditionДокумент36 страницTest Bank For The Psychology of Health and Health Care A Canadian Perspective 5th Editionload.notablewp0oz100% (37)
- How To Add Attachment Using JAVA MappingДокумент4 страницыHow To Add Attachment Using JAVA MappingmvrooyenОценок пока нет
- DCN Dte-Dce and ModemsДокумент5 страницDCN Dte-Dce and ModemsSathish BabuОценок пока нет
- SDN Van NotesДокумент26 страницSDN Van Notesmjsmith11Оценок пока нет
- Ancient Sumer Flip BookДокумент9 страницAncient Sumer Flip Bookapi-198624210Оценок пока нет
- Bgrim 1q2022Документ56 страницBgrim 1q2022Dianne SabadoОценок пока нет
- Amritsar Police StationДокумент5 страницAmritsar Police StationRashmi KbОценок пока нет
- Literature Review of Service Quality in RestaurantsДокумент7 страницLiterature Review of Service Quality in RestaurantsuifjzvrifОценок пока нет
- Data Network Unit 6 - UCДокумент15 страницData Network Unit 6 - UCANISHA DONDEОценок пока нет
- Cpar Characteristics and Functions Week 3Документ128 страницCpar Characteristics and Functions Week 3christianwood0117Оценок пока нет
- EQ JOURNAL 2 - AsioДокумент3 страницыEQ JOURNAL 2 - AsioemanОценок пока нет