Вам также может понравиться
- Starship Troopers Skinnies Army BookДокумент66 страницStarship Troopers Skinnies Army BookDougBirtles100% (5)
- Drug Discovery and DevelopmentДокумент28 страницDrug Discovery and DevelopmentMirza Shaharyar BaigОценок пока нет
- Drug DesignДокумент19 страницDrug DesignnitishpathaniaОценок пока нет
- Food Processing Technology I: Defry Lesmana, M.K.M. Aprilia Fitriani, M.SCДокумент29 страницFood Processing Technology I: Defry Lesmana, M.K.M. Aprilia Fitriani, M.SCAprilia Oanima100% (1)
- A Handbook of Mcqs in Toxicology: Muneeb U RehmanДокумент53 страницыA Handbook of Mcqs in Toxicology: Muneeb U Rehmanquimicosorio100% (2)
- Biosensors PowerpointДокумент26 страницBiosensors PowerpointDevansh DurgarajuОценок пока нет
- An Introduction to Mechanisms in Pharmacology and TherapeuticsОт EverandAn Introduction to Mechanisms in Pharmacology and TherapeuticsОценок пока нет
- Chiro Guidelines WHOДокумент51 страницаChiro Guidelines WHOMinh MinhОценок пока нет
- The Modification of Natural ProductsДокумент18 страницThe Modification of Natural ProductsElena GonzálezОценок пока нет
- Microbiological Test of Medical DevicesДокумент5 страницMicrobiological Test of Medical Devicesbijendra_sinhaОценок пока нет
- Mechanobiology and Diseases of Mechanotransduction: Donald E IngberДокумент14 страницMechanobiology and Diseases of Mechanotransduction: Donald E IngberTezar AndreanОценок пока нет
- Pharmacology in Drug Discovery: Understanding Drug ResponseОт EverandPharmacology in Drug Discovery: Understanding Drug ResponseОценок пока нет
- Questions and Answers For Scientific Article June 2011Документ14 страницQuestions and Answers For Scientific Article June 2011Zydhan ZahidОценок пока нет
- Article 1609836991Документ9 страницArticle 1609836991Read WhiteОценок пока нет
- Pelepasan Polimer LangerДокумент7 страницPelepasan Polimer LangerUntia Kartika Sari RamadhaniОценок пока нет
- Pharmacology PrelimДокумент43 страницыPharmacology PrelimJohn CarloОценок пока нет
- Revi SysteДокумент20 страницRevi Systedr.israquiОценок пока нет
- New Drugs ArtritisДокумент13 страницNew Drugs ArtritisqfecrespoОценок пока нет
- PHA3801 Short Answer QuestionsДокумент5 страницPHA3801 Short Answer QuestionsFarhana Azmira AsmadiОценок пока нет
- J. Nucl. Med. Technol. 2018 Currie Jnmt.117.199588Документ19 страницJ. Nucl. Med. Technol. 2018 Currie Jnmt.117.199588Mi MeaОценок пока нет
- Pdynamics For PrescrberДокумент6 страницPdynamics For PrescrberpdladvaОценок пока нет
- Environmental ImpactsДокумент10 страницEnvironmental ImpactsIndra DwinataОценок пока нет
- Biomedical Application of Enzyme ReviewДокумент18 страницBiomedical Application of Enzyme Reviewኢትዮ-360 ሜድያОценок пока нет
- Introduction To Protein Folding For Physicists: 1 Why Study Proteins?Документ53 страницыIntroduction To Protein Folding For Physicists: 1 Why Study Proteins?Alishba Faixan100% (1)
- Drug Designing, Discovery and Development Techniques: Elvis A. Martis and Rakesh R. SomaniДокумент19 страницDrug Designing, Discovery and Development Techniques: Elvis A. Martis and Rakesh R. SomanitahafeОценок пока нет
- The Potential Action of Ssris in The Treatment of Skin Diseases Including Atopic Dermatitis and Slow Healing WoundsДокумент9 страницThe Potential Action of Ssris in The Treatment of Skin Diseases Including Atopic Dermatitis and Slow Healing WoundsJosé Luis MacedoОценок пока нет
- Biomedical ScienceДокумент8 страницBiomedical ScienceBiology BảoОценок пока нет
- Li Weber2013Документ9 страницLi Weber2013Hector Javier BurgosОценок пока нет
- Epothilones: Mechanism of Action and Biologic Activity: Susan Goodin, Michael P. Kane, and Eric H. RubinДокумент12 страницEpothilones: Mechanism of Action and Biologic Activity: Susan Goodin, Michael P. Kane, and Eric H. Rubinmuhammad_taufik_mОценок пока нет
- Answers by Stafford Valentine Redden, VIHS, Head of Biology 1Документ14 страницAnswers by Stafford Valentine Redden, VIHS, Head of Biology 1areyouthere92Оценок пока нет
- Chemical GeneticsДокумент10 страницChemical GeneticsMatei BuneaОценок пока нет
- Cell SELEX ProcedureДокумент11 страницCell SELEX ProcedurenamgeshОценок пока нет
- "Science" Editor's Choice "A Survey of Entrepreneurial Risk in Stream and Compensatory Mitigation Markets"Документ2 страницы"Science" Editor's Choice "A Survey of Entrepreneurial Risk in Stream and Compensatory Mitigation Markets"Restoration Systems, LLCОценок пока нет
- Nats 1675Документ5 страницNats 1675Tanveer KhuranaОценок пока нет
- Bioorganic & Medicinal ChemistryДокумент14 страницBioorganic & Medicinal ChemistryAulia Fatmiyatun El-FalesyОценок пока нет
- N Asmita Bandyopadhyay 2233645 - Cia IДокумент10 страницN Asmita Bandyopadhyay 2233645 - Cia IN ASMITA BANDYOPADHYAY 2233645Оценок пока нет
- In Silico Pharmacology For Drug Discovery: Methods For Virtual Ligand Screening and ProfilingДокумент12 страницIn Silico Pharmacology For Drug Discovery: Methods For Virtual Ligand Screening and ProfilingDwi PuspitaОценок пока нет
- Fortune Vushe CH205 Assignment 1Документ4 страницыFortune Vushe CH205 Assignment 1Fortune VusheОценок пока нет
- Pharmaceuticals 14 00984Документ23 страницыPharmaceuticals 14 00984Walid Ebid ElgammalОценок пока нет
- Literature Review On Oxidative StressДокумент4 страницыLiterature Review On Oxidative Stressc5p8vze7100% (1)
- New Approaches To Treat Cancer - What They Can and Cannot DoДокумент10 страницNew Approaches To Treat Cancer - What They Can and Cannot DoMuhammad ImranОценок пока нет
- Stewart 2016Документ4 страницыStewart 2016alan.rangel.puenteОценок пока нет
- Dr. Doni Priambodo, SP - PD-KPTI, FINASIM - Immunomodulator and Antioxidant in SepsisДокумент22 страницыDr. Doni Priambodo, SP - PD-KPTI, FINASIM - Immunomodulator and Antioxidant in SepsisOlivia DwimaswastiОценок пока нет
- JNP16 691Документ6 страницJNP16 691Carolina Cumbicus TorresОценок пока нет
- JCM 11 02614 v3Документ9 страницJCM 11 02614 v3Jimmy MendozaОценок пока нет
- Chem - Organic SynthesisДокумент8 страницChem - Organic SynthesiswhoyaОценок пока нет
- Adjuvant Mechanism Paper PDFДокумент10 страницAdjuvant Mechanism Paper PDFAnand Prakash YadavОценок пока нет
- Immunosupresive AgentДокумент12 страницImmunosupresive AgentTamam JauharОценок пока нет
- HHS Public Access: Epigenetic Polypharmacology: A New Frontier For Epi-Drug DiscoveryДокумент76 страницHHS Public Access: Epigenetic Polypharmacology: A New Frontier For Epi-Drug DiscoveryDANIELA TOMASELLIОценок пока нет
- Pharmacology IДокумент82 страницыPharmacology IMelanieОценок пока нет
- Drugs For Chemical EngineeringДокумент34 страницыDrugs For Chemical Engineeringshivakumar hrОценок пока нет
- Echinochrome A and Cytokine Storm SyndromeДокумент11 страницEchinochrome A and Cytokine Storm SyndromeNicolas Fernandez RubilarОценок пока нет
- marinedrugsДокумент20 страницmarinedrugsreni vionitaОценок пока нет
- Protection For Medication Induced Hearing Loss The State of The ScienceДокумент10 страницProtection For Medication Induced Hearing Loss The State of The ScienceTika Renwarin Tua ElОценок пока нет
- Proof: Informatics in Medicine UnlockedДокумент8 страницProof: Informatics in Medicine UnlockedNia PortillaОценок пока нет
- Introduction To PkokineticsДокумент3 страницыIntroduction To PkokineticspdladvaОценок пока нет
- General-Pharmacology TermsДокумент17 страницGeneral-Pharmacology TermsSachin ParamashettiОценок пока нет
- Medicinal Chemistry Dissertation TopicsДокумент6 страницMedicinal Chemistry Dissertation TopicsWriteMyPaperPleaseNewHaven100% (1)
- Immunosuppression and Kidney TrnsplantДокумент25 страницImmunosuppression and Kidney TrnsplantAbdul QuyyumОценок пока нет
- Pathophysiology of Anorexia in The Cancer Cachexia Syndrome: Chukwuemeka Charles Ezeoke & John E. MorleyДокумент16 страницPathophysiology of Anorexia in The Cancer Cachexia Syndrome: Chukwuemeka Charles Ezeoke & John E. Morleydevin mahendikaОценок пока нет
- EnzymeДокумент5 страницEnzymeSepta DewiОценок пока нет
- Alzheimer Disease New TreatmentsДокумент22 страницыAlzheimer Disease New TreatmentsDaniela BlancoОценок пока нет
- (Pangfung) Bioactive CarbohydrateДокумент42 страницы(Pangfung) Bioactive CarbohydrateAprilia OanimaОценок пока нет
- Sago PalmДокумент317 страницSago PalmAprilia OanimaОценок пока нет
- Catalytic Mechanism of Angiotensin-Converting Enzyme and Effects Chloride IonДокумент11 страницCatalytic Mechanism of Angiotensin-Converting Enzyme and Effects Chloride IonAprilia OanimaОценок пока нет
- Brain Damage in Infant Mice Following Oral Intake of Glutamate, Aspartate, and Cystein (Olney 1970)Документ3 страницыBrain Damage in Infant Mice Following Oral Intake of Glutamate, Aspartate, and Cystein (Olney 1970)Aprilia OanimaОценок пока нет
- Jams, Jellies and Marmalades: Raw MaterialsДокумент39 страницJams, Jellies and Marmalades: Raw MaterialsHafiz Abu BakarОценок пока нет
- Brain Lesions, Obesity, and Other Disturbances in Mice Treated With Monosodium Glutamate (Olney 1969)Документ3 страницыBrain Lesions, Obesity, and Other Disturbances in Mice Treated With Monosodium Glutamate (Olney 1969)Aprilia OanimaОценок пока нет
- Crystal Structure of The Human ACE - Lisonopril ComplexДокумент4 страницыCrystal Structure of The Human ACE - Lisonopril ComplexAprilia OanimaОценок пока нет
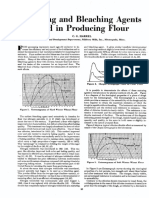
- Used Producing Flour: Maturing and Bleaching AgentsДокумент6 страницUsed Producing Flour: Maturing and Bleaching AgentsAprilia OanimaОценок пока нет
- Angiotensin-I-converting Enzyme and Its RelativesДокумент5 страницAngiotensin-I-converting Enzyme and Its RelativesAprilia OanimaОценок пока нет
- Angiotensin-Converting Enzyme 2 (ACE2) Is A Key Modulator of RASДокумент9 страницAngiotensin-Converting Enzyme 2 (ACE2) Is A Key Modulator of RASAprilia OanimaОценок пока нет
- Non Thermal PreservativeДокумент56 страницNon Thermal PreservativeAprilia OanimaОценок пока нет
- Teknologi Pengolahan Pangan - Raw MaterialДокумент44 страницыTeknologi Pengolahan Pangan - Raw MaterialAprilia OanimaОценок пока нет
- Food Processing Technology I: Aprilia Fitriani, M.Sc. Defry Lesmana, M.K.MДокумент22 страницыFood Processing Technology I: Aprilia Fitriani, M.Sc. Defry Lesmana, M.K.MAprilia OanimaОценок пока нет
- Food Material Science: Efendi Oulan Gustav Hakim Nata Buana, S.TP. M.Eng. Aprilia Fitriani, S.TP., M.SCДокумент22 страницыFood Material Science: Efendi Oulan Gustav Hakim Nata Buana, S.TP. M.Eng. Aprilia Fitriani, S.TP., M.SCAprilia OanimaОценок пока нет
- Cooking Fruits and VegetablesДокумент26 страницCooking Fruits and VegetablesAprilia OanimaОценок пока нет
- Food Processing Technology I: Defry Lesmana, M.K.M. Aprilia Fitriani, M.SCДокумент37 страницFood Processing Technology I: Defry Lesmana, M.K.M. Aprilia Fitriani, M.SCAprilia OanimaОценок пока нет
- (Anpang) Moisture AnalysisДокумент46 страниц(Anpang) Moisture AnalysisAprilia OanimaОценок пока нет
- (KBP) Density and Specific GravityДокумент74 страницы(KBP) Density and Specific GravityAprilia OanimaОценок пока нет
- (Anpang) Lipid AnalysisДокумент53 страницы(Anpang) Lipid AnalysisAprilia OanimaОценок пока нет
- Teknologi Pengolahan Pangan - Raw MaterialДокумент44 страницыTeknologi Pengolahan Pangan - Raw MaterialAprilia OanimaОценок пока нет
- (Anpang) Carbohydrate AnalysisДокумент29 страниц(Anpang) Carbohydrate AnalysisAprilia OanimaОценок пока нет
- Lab Exercise 0ne: Carbohydrate Analysis Lab A.1 (Page 28)Документ31 страницаLab Exercise 0ne: Carbohydrate Analysis Lab A.1 (Page 28)Goh Kae HorngОценок пока нет
- Comparison of the Fully Automated Urinalysis Analyzers UX-2000 and Cobas 6500Документ10 страницComparison of the Fully Automated Urinalysis Analyzers UX-2000 and Cobas 6500Sethulakshmi PharmacistОценок пока нет
- 1oksidative Stress TestДокумент34 страницы1oksidative Stress TestDian WijayantiОценок пока нет
- Science Holidays Homework Class 9Документ2 страницыScience Holidays Homework Class 9nileshОценок пока нет
- 2010 FulltextДокумент410 страниц2010 FulltextNajam Din SahitoОценок пока нет
- Sample Lab ReportДокумент7 страницSample Lab ReportPutri Syalieyana0% (1)
- Diencephalon PresentationДокумент19 страницDiencephalon Presentation300rОценок пока нет
- Module 2 NCM 114Документ11 страницModule 2 NCM 114Erven AranasОценок пока нет
- Citric Acid Production Stirred TankДокумент9 страницCitric Acid Production Stirred TankKarliiux MedinaОценок пока нет
- Forest Health and Biotechnology - Possibilities and Considerations (2019)Документ241 страницаForest Health and Biotechnology - Possibilities and Considerations (2019)apertusОценок пока нет
- Ewh Ix PDFДокумент80 страницEwh Ix PDFOR Premium FreeОценок пока нет
- Full Essay Academic SkillsДокумент3 страницыFull Essay Academic SkillsNurl AinaОценок пока нет
- Thesis Olof HerttingДокумент57 страницThesis Olof HerttingAjay IyerОценок пока нет
- Physiology of Cerebellum Lec Foe 2nd Year by DR Sadia Uploaded by ZaighamДокумент61 страницаPhysiology of Cerebellum Lec Foe 2nd Year by DR Sadia Uploaded by ZaighamZaigham HammadОценок пока нет
- PMLSP 2 ReviewerДокумент38 страницPMLSP 2 ReviewerSophia Mae ClavecillaОценок пока нет
- (PDF) Root Growth of Phalsa (Grewia Asiatica L.) As Affected by Type of CuttДокумент5 страниц(PDF) Root Growth of Phalsa (Grewia Asiatica L.) As Affected by Type of CuttAliОценок пока нет
- Natamycin Story - What You Need to KnowДокумент13 страницNatamycin Story - What You Need to KnowCharles MardiniОценок пока нет
- Botany AssignmentДокумент35 страницBotany AssignmentLakshmiОценок пока нет
- Physiological Psychology ReviewerДокумент13 страницPhysiological Psychology ReviewerLeopando Rod CyrenzОценок пока нет
- Adolescent Reproductive and Sexual HealthДокумент42 страницыAdolescent Reproductive and Sexual HealthMuhammad Abbas WaliОценок пока нет
- Met. Secundarios Cáscara SagradaДокумент9 страницMet. Secundarios Cáscara SagradaDalíAsesoríasОценок пока нет
- Amazing Facts - Exercises 1Документ3 страницыAmazing Facts - Exercises 1Alina DorobatОценок пока нет
- General Biology 2 Budget of WorkДокумент3 страницыGeneral Biology 2 Budget of WorkMaricris BalboaОценок пока нет
- Pengantar Mikrobiologi LingkunganДокумент28 страницPengantar Mikrobiologi LingkunganAhmad SyahrulОценок пока нет
- Student Exploration: Building DNAДокумент4 страницыStudent Exploration: Building DNAMia Smith100% (1)
- Young Children's Biological Predisposition To Learn in Privileged DomainДокумент6 страницYoung Children's Biological Predisposition To Learn in Privileged DomainVeronica Dadal0% (1)
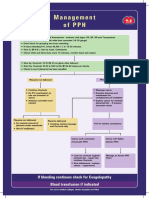
- Management of PPHДокумент1 страницаManagement of PPH098 U.KARTHIK SARAVANA KANTHОценок пока нет